

Global patterns and rates of habitat transitions across the eukaryotic tree of life
- PMID: 35927316
- PMCID: PMC9525238
- DOI: 10.1038/s41559-022-01838-4
Global patterns and rates of habitat transitions across the eukaryotic tree of life
Abstract
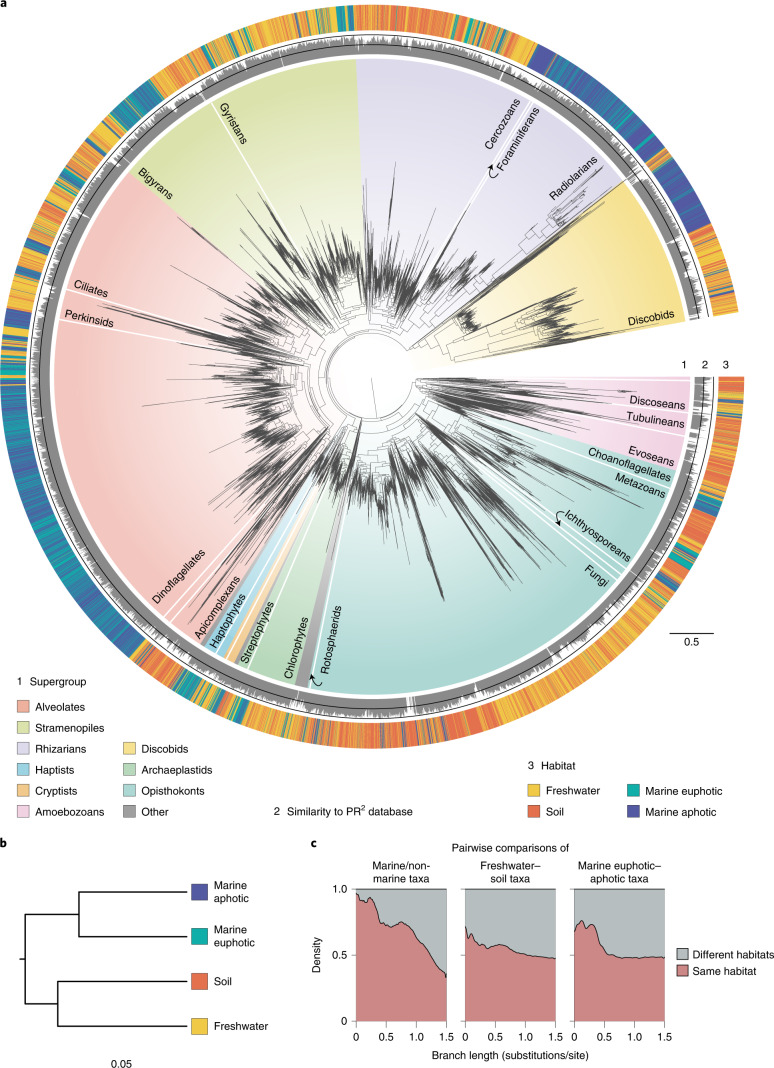
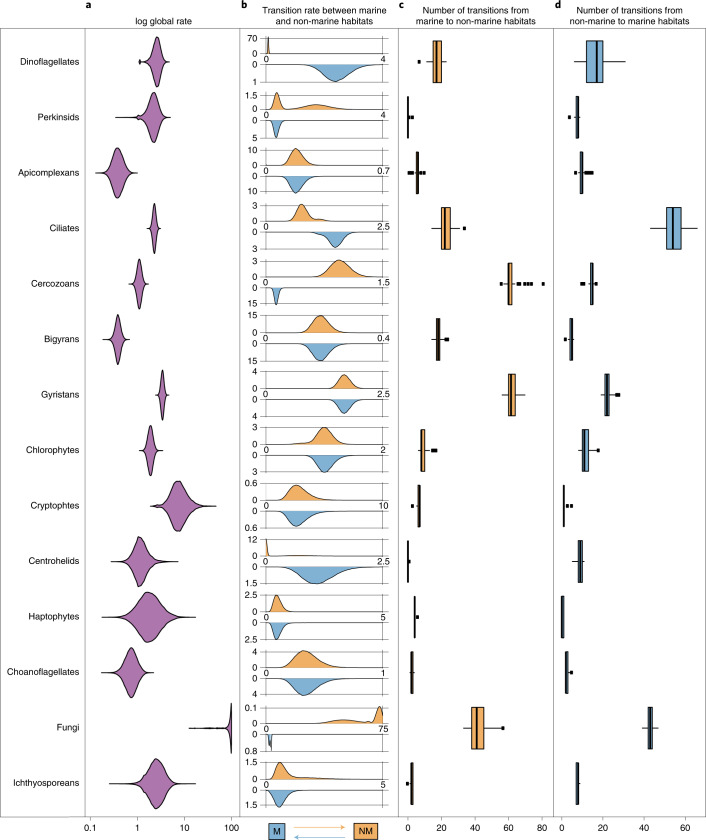
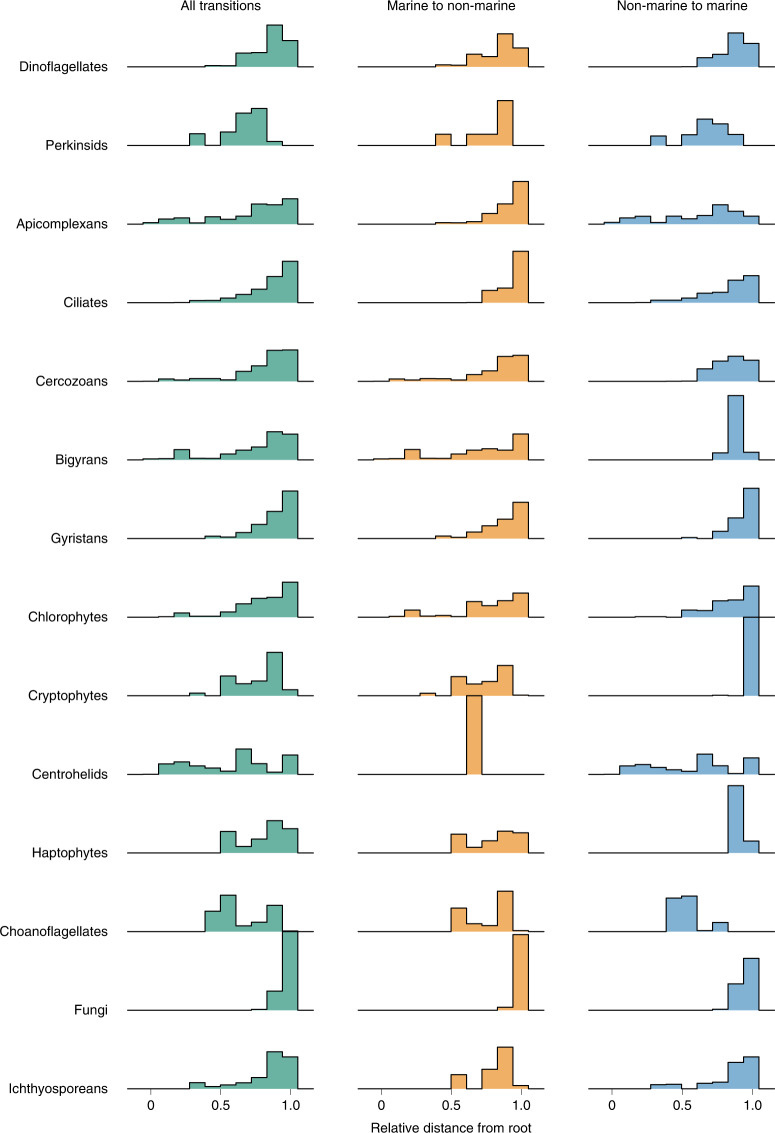
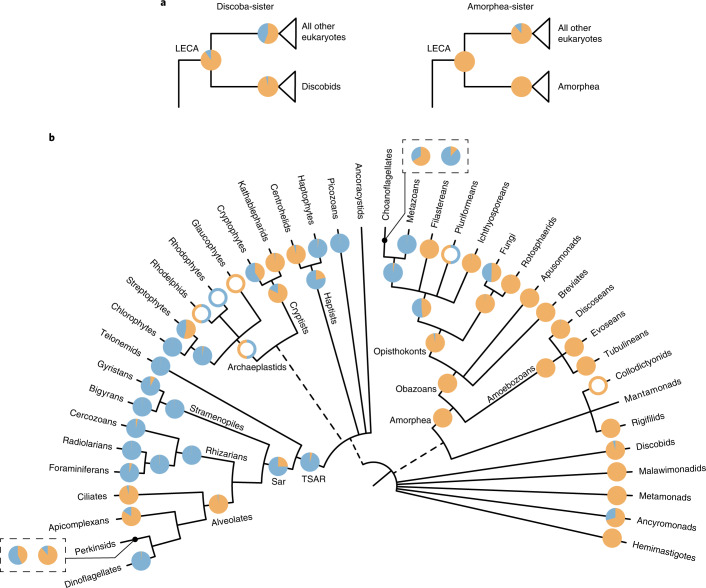
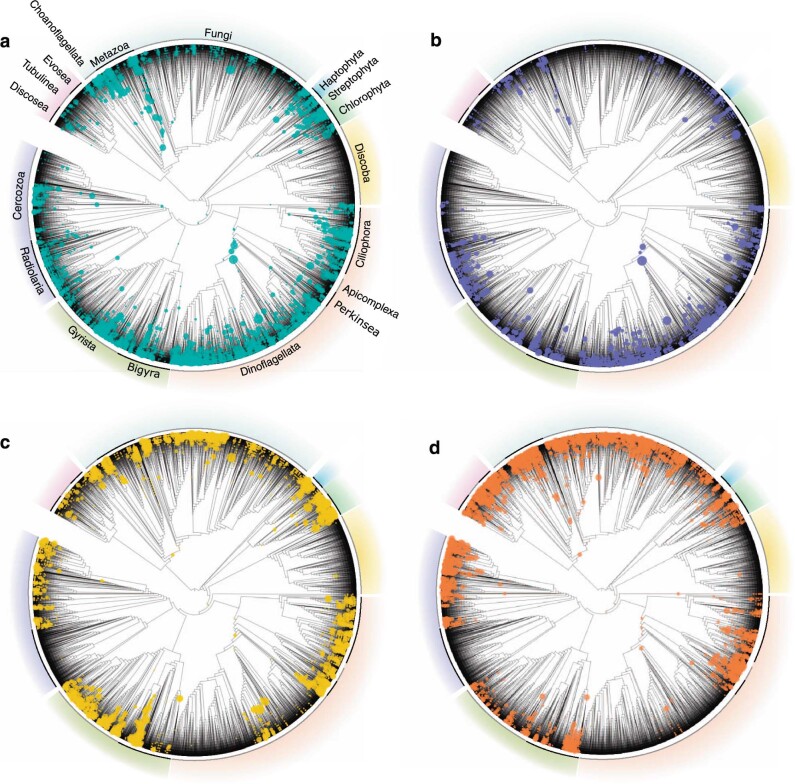
The successful colonization of new habitats has played a fundamental role during the evolution of life. Salinity is one of the strongest barriers for organisms to cross, which has resulted in the evolution of distinct marine and non-marine (including both freshwater and soil) communities. Although microbes represent by far the vast majority of eukaryote diversity, the role of the salt barrier in shaping the diversity across the eukaryotic tree is poorly known. Traditional views suggest rare and ancient marine/non-marine transitions but this view is being challenged by the discovery of several recently transitioned lineages. Here, we investigate habitat evolution across the tree of eukaryotes using a unique set of taxon-rich phylogenies inferred from a combination of long-read and short-read environmental metabarcoding data spanning the ribosomal DNA operon. Our results show that, overall, marine and non-marine microbial communities are phylogenetically distinct but transitions have occurred in both directions in almost all major eukaryotic lineages, with hundreds of transition events detected. Some groups have experienced relatively high rates of transitions, most notably fungi for which crossing the salt barrier has probably been an important aspect of their successful diversification. At the deepest phylogenetic levels, ancestral habitat reconstruction analyses suggest that eukaryotes may have first evolved in non-marine habitats and that the two largest known eukaryotic assemblages (TSAR and Amorphea) arose in different habitats. Overall, our findings indicate that the salt barrier has played an important role during eukaryote evolution and provide a global perspective on habitat transitions in this domain of life.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Molecular phylogenetics supports multiple evolutionary transitions from marine to freshwater habitats in ariid catfishes.Mol Phylogenet Evol. 2010 Apr;55(1):249-258. doi: 10.1016/j.ympev.2009.12.018. Epub 2010 Jan 4. Mol Phylogenet Evol. 2010. PMID: 20045737
-
Diversification of unicellular eukaryotes: cryptomonad colonizations of marine and fresh waters inferred from revised 18S rRNA phylogeny.Environ Microbiol. 2008 Oct;10(10):2635-44. doi: 10.1111/j.1462-2920.2008.01685.x. Epub 2008 Jul 16. Environ Microbiol. 2008. PMID: 18643928
-
Reevaluating the Salty Divide: Phylogenetic Specificity of Transitions between Marine and Freshwater Systems.mSystems. 2018 Nov 13;3(6):e00232-18. doi: 10.1128/mSystems.00232-18. eCollection 2018 Nov-Dec. mSystems. 2018. PMID: 30443603 Free PMC article.
-
Unveiling new microbial eukaryotes in the surface ocean.Curr Opin Microbiol. 2008 Jun;11(3):213-8. doi: 10.1016/j.mib.2008.04.004. Epub 2008 Jun 13. Curr Opin Microbiol. 2008. PMID: 18556239 Review.
-
The phagotrophic origin of eukaryotes and phylogenetic classification of Protozoa.Int J Syst Evol Microbiol. 2002 Mar;52(Pt 2):297-354. doi: 10.1099/00207713-52-2-297. Int J Syst Evol Microbiol. 2002. PMID: 11931142 Review.
Cited by
-
Long-Term Investigation of Marine-Derived Aspergillus Diversity in the Republic of Korea.Mycobiology. 2023 Dec 27;51(6):436-444. doi: 10.1080/12298093.2023.2279342. eCollection 2023. Mycobiology. 2023. PMID: 38179120 Free PMC article.
-
Population genomic analyses reveal that salinity and geographic isolation drive diversification in a free-living protist.Sci Rep. 2024 Feb 29;14(1):4986. doi: 10.1038/s41598-024-55362-5. Sci Rep. 2024. PMID: 38424140 Free PMC article.
-
Who are the marine fungi?Environ Microbiol. 2023 Jan;25(1):131-134. doi: 10.1111/1462-2920.16240. Epub 2022 Oct 23. Environ Microbiol. 2023. PMID: 36217720 Free PMC article. No abstract available.
-
Convergent Adaptation of True Crabs (Decapoda: Brachyura) to a Gradient of Terrestrial Environments.Syst Biol. 2024 Jul 27;73(2):247-262. doi: 10.1093/sysbio/syad066. Syst Biol. 2024. PMID: 37941464 Free PMC article.
-
The Ribosomal Operon Database: A Full-Length rDNA Operon Database Derived From Genome Assemblies.Mol Ecol Resour. 2025 Jan;25(1):e14031. doi: 10.1111/1755-0998.14031. Epub 2024 Oct 21. Mol Ecol Resour. 2025. PMID: 39428982 Free PMC article.
References
-
- Simpson, G. G. The Major Features of Evolution (Columbia Univ. Press, 1953).
-
- Osborn HF. The law of adaptive radiation. Am. Nat. 1902;36:353–363. doi: 10.1086/278137. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
