

Molecular map of chronic lymphocytic leukemia and its impact on outcome
- PMID: 35927489
- PMCID: PMC10084830
- DOI: 10.1038/s41588-022-01140-w
Molecular map of chronic lymphocytic leukemia and its impact on outcome
Abstract
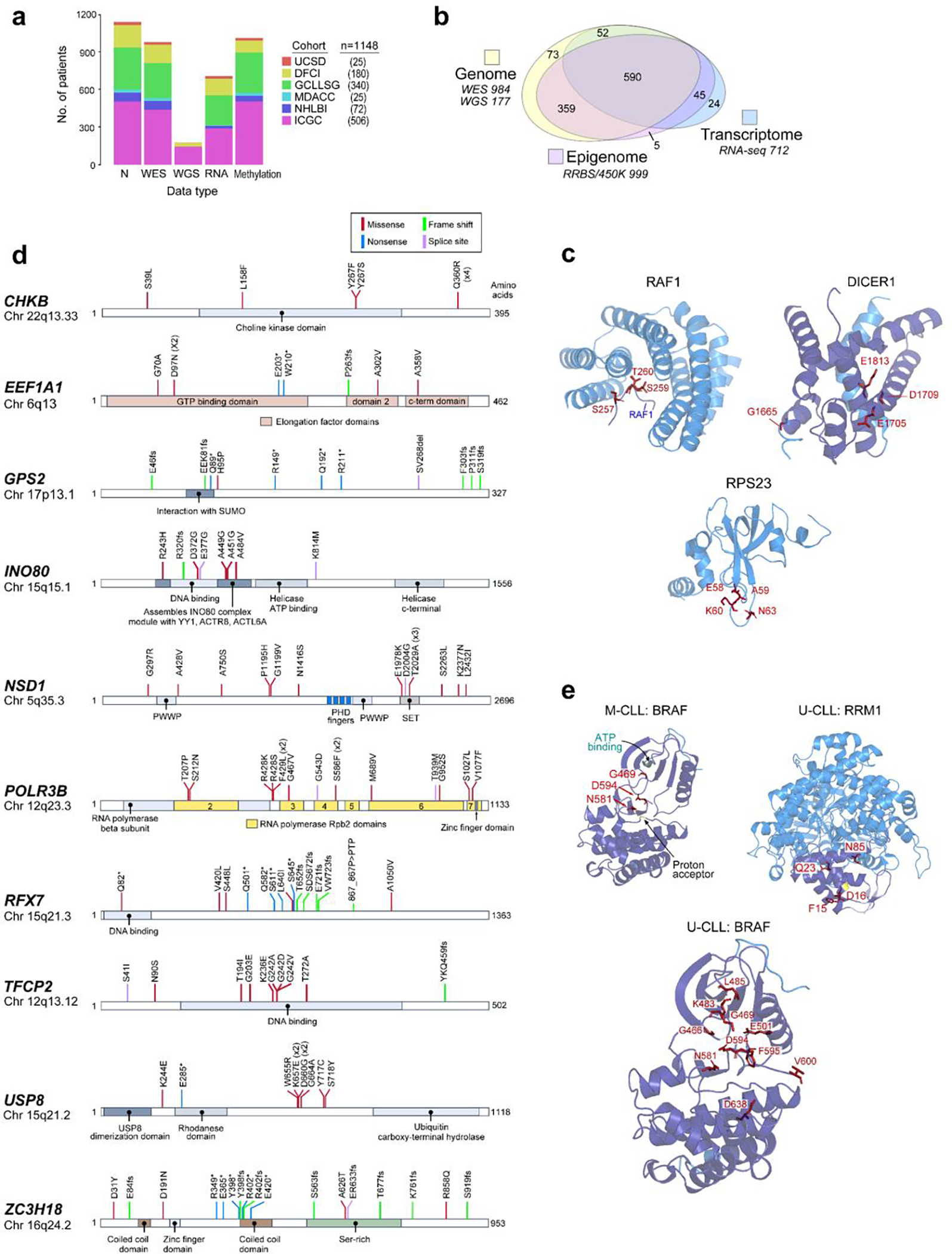
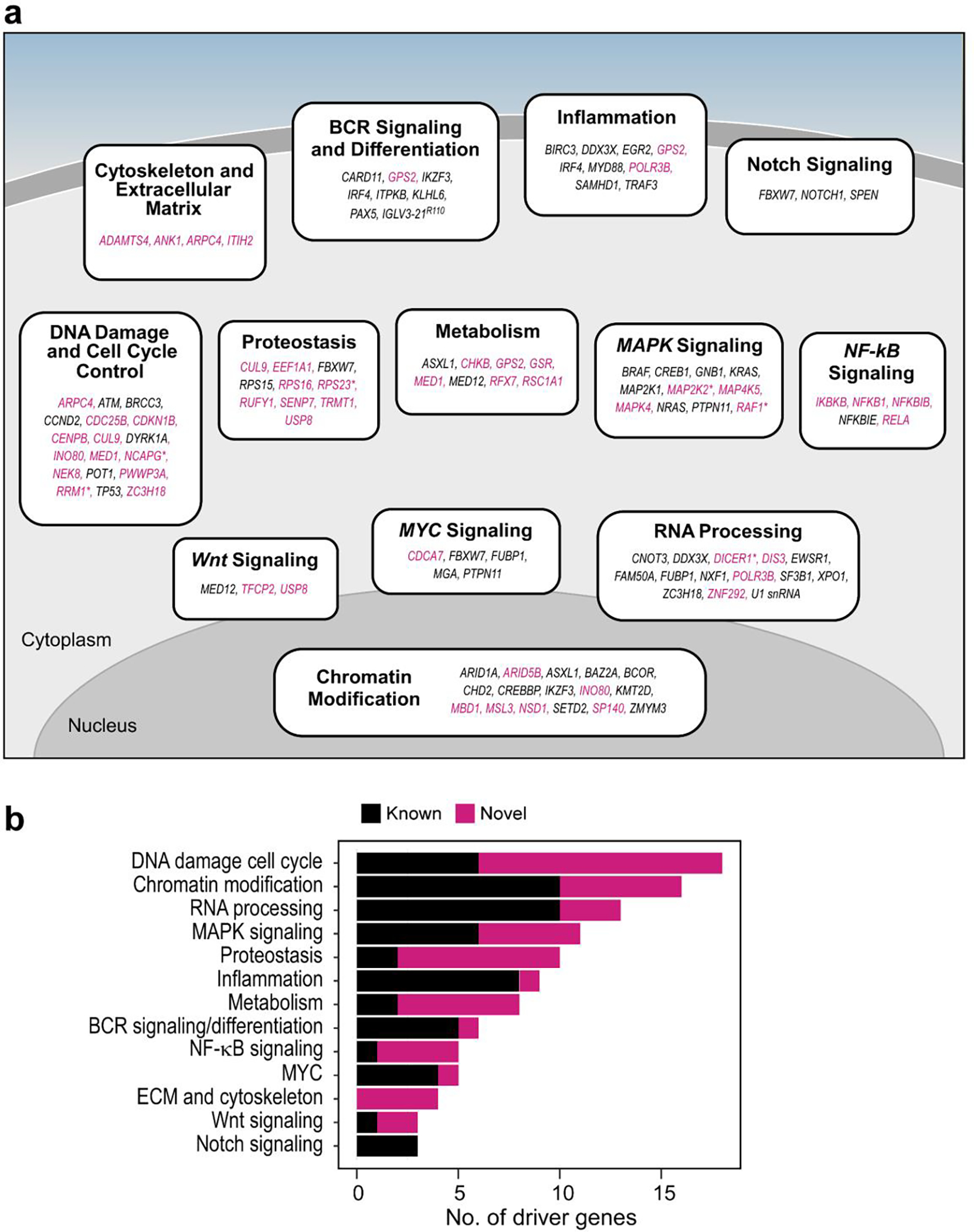
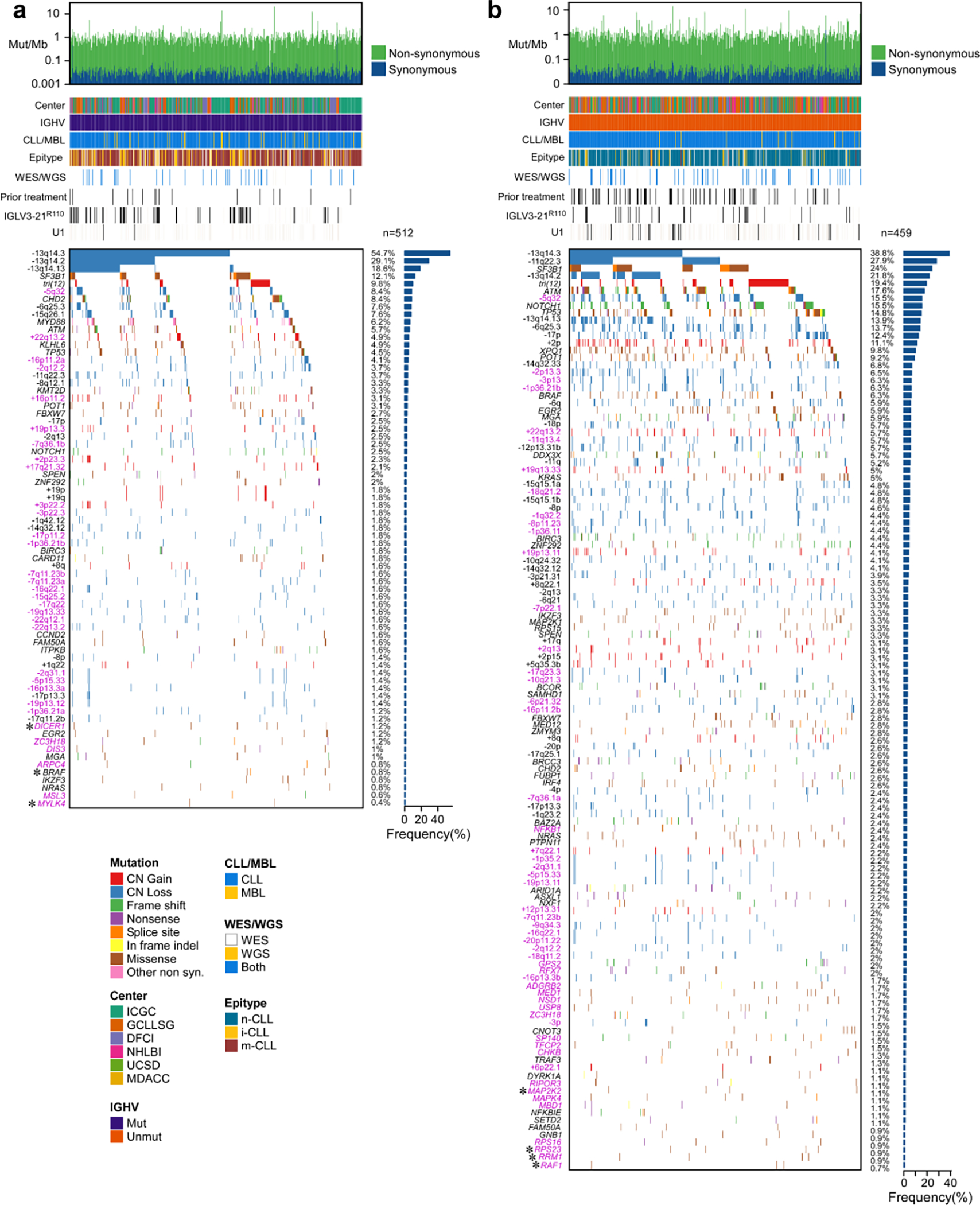
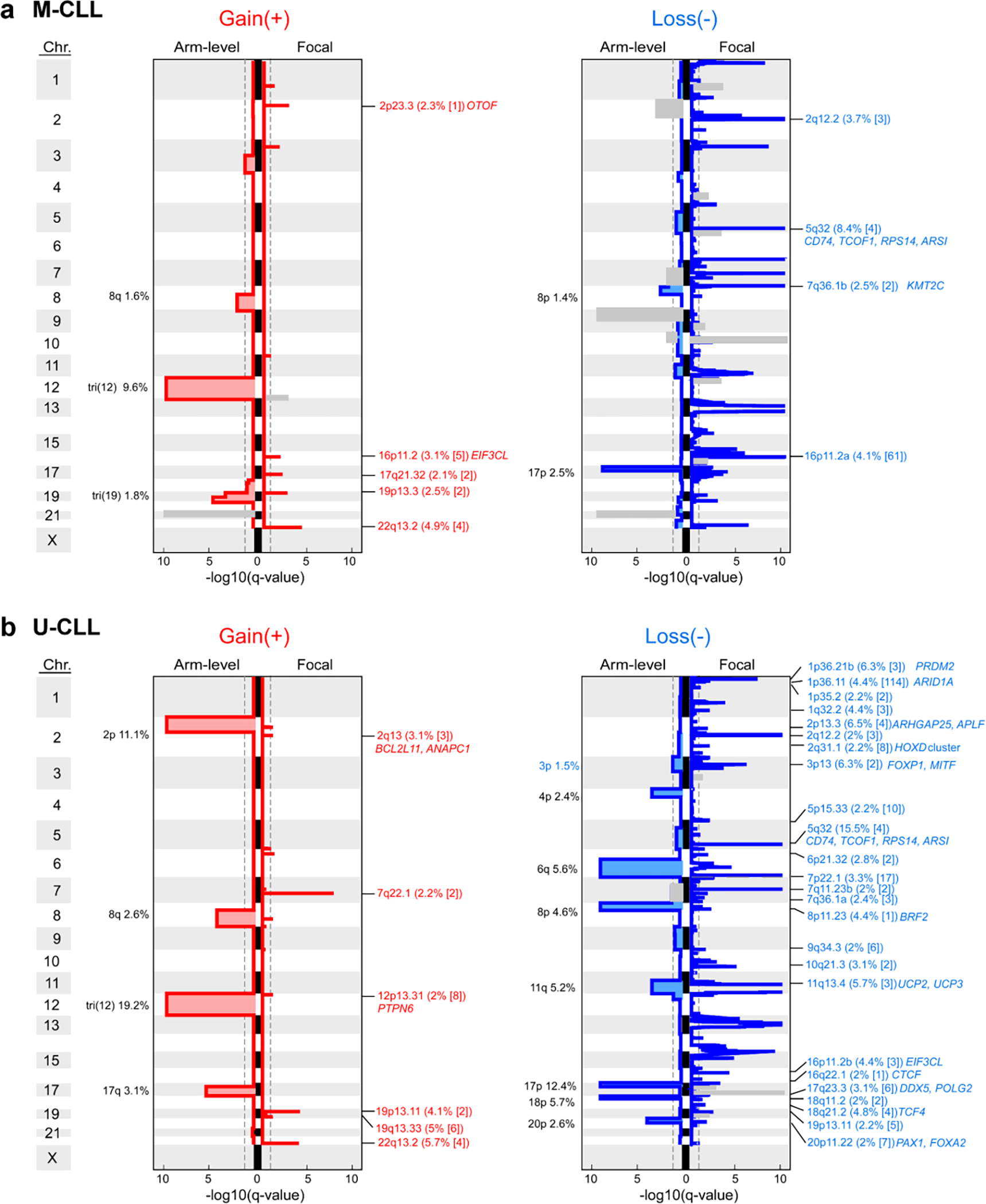
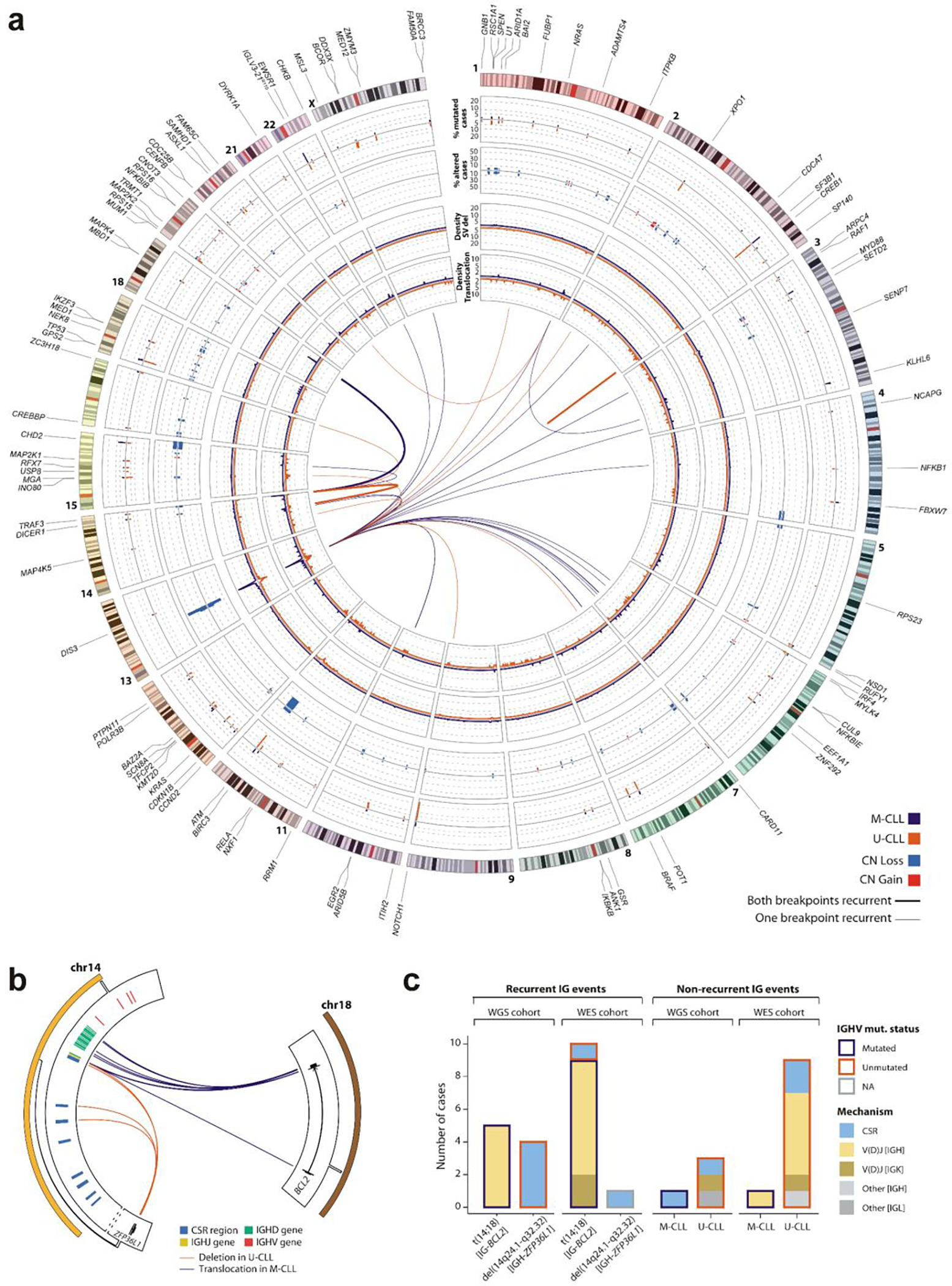
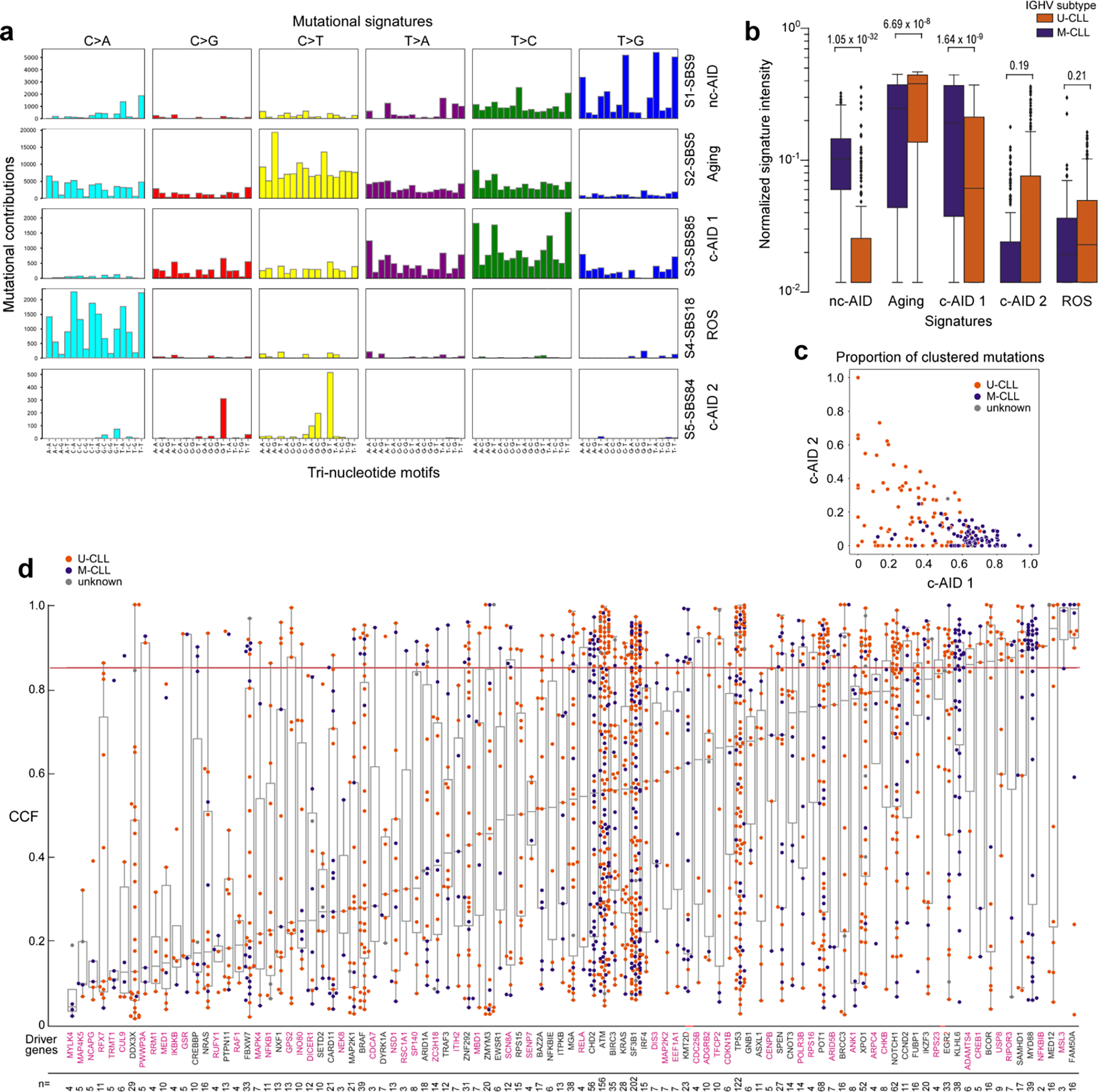
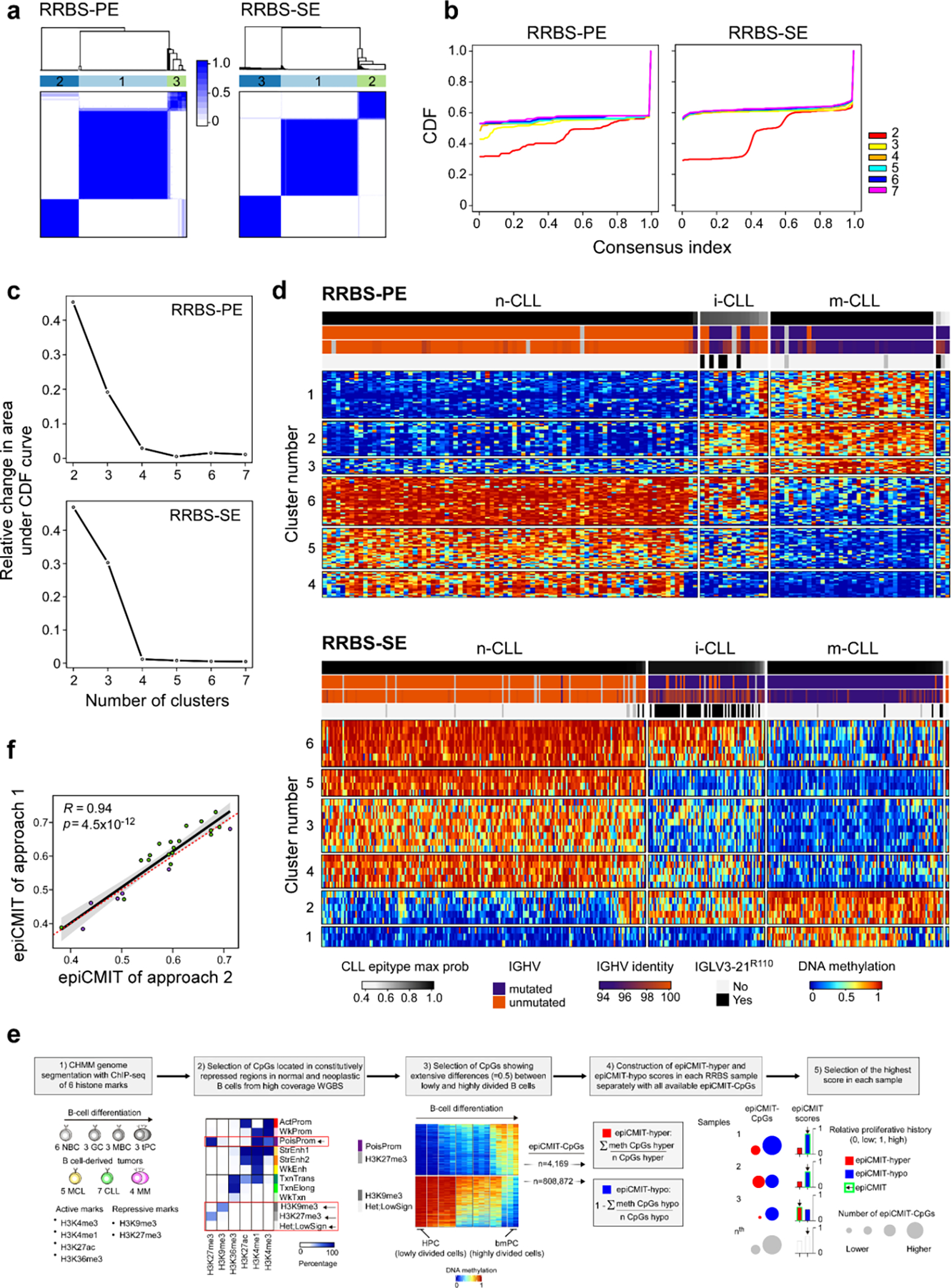
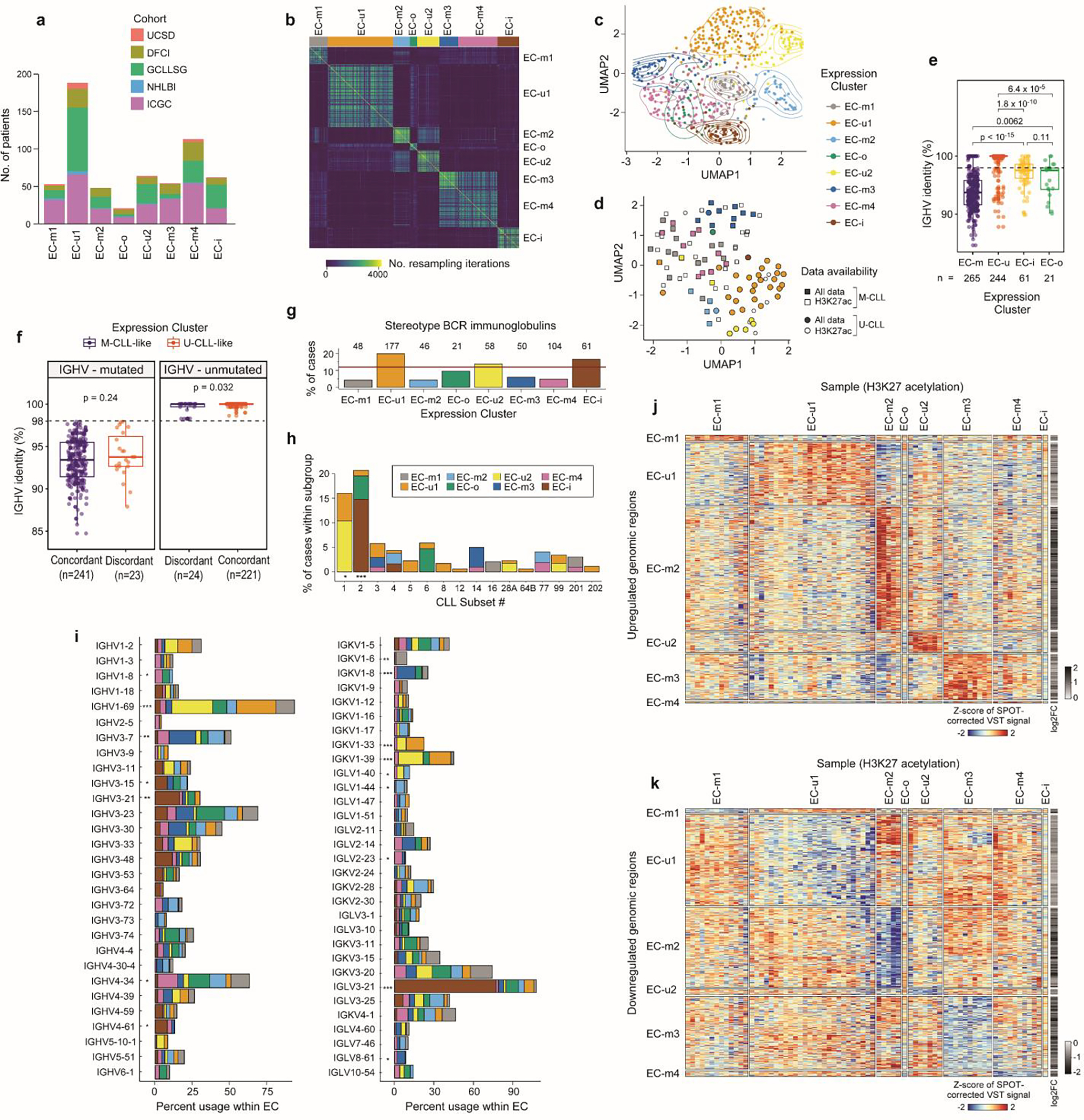
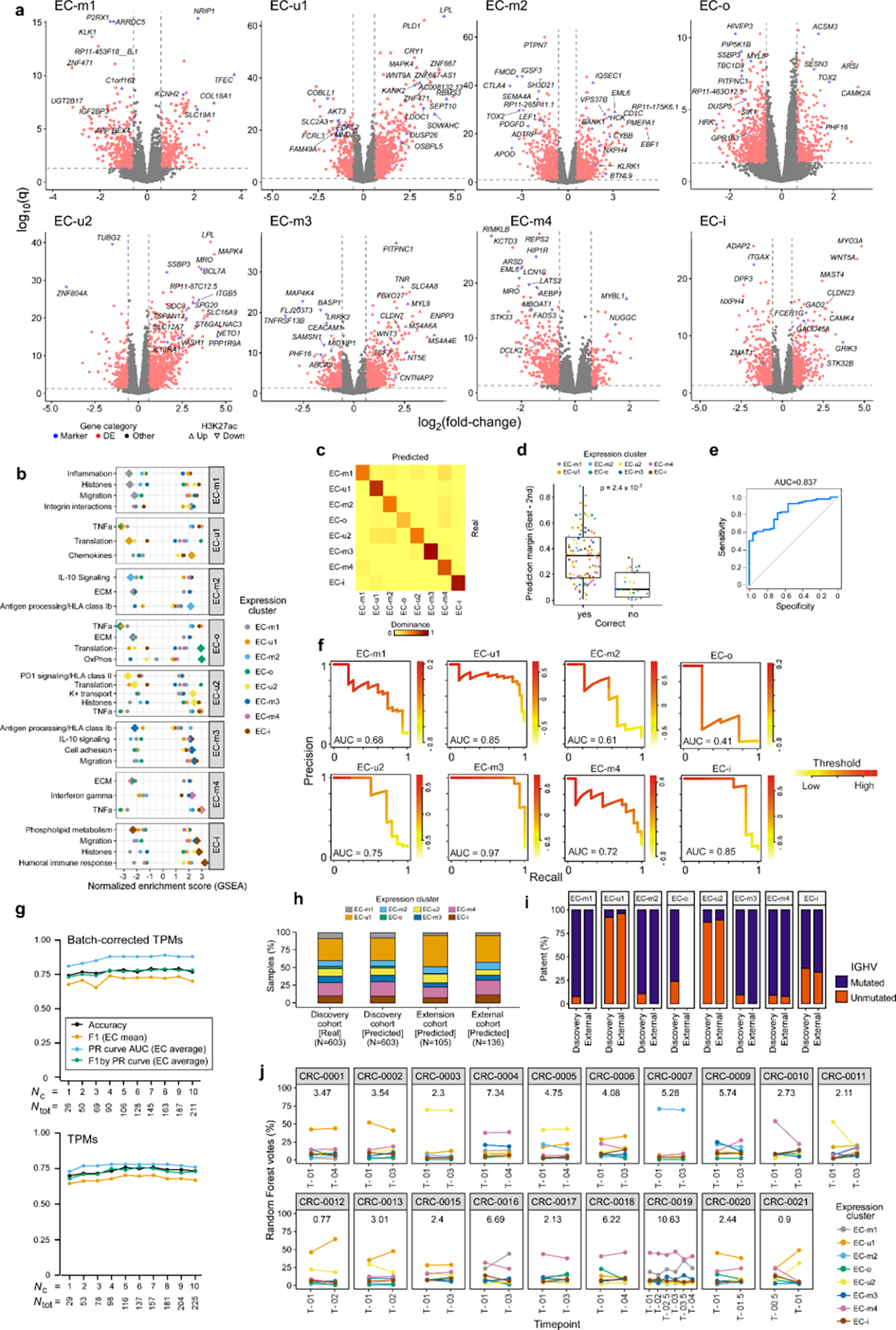
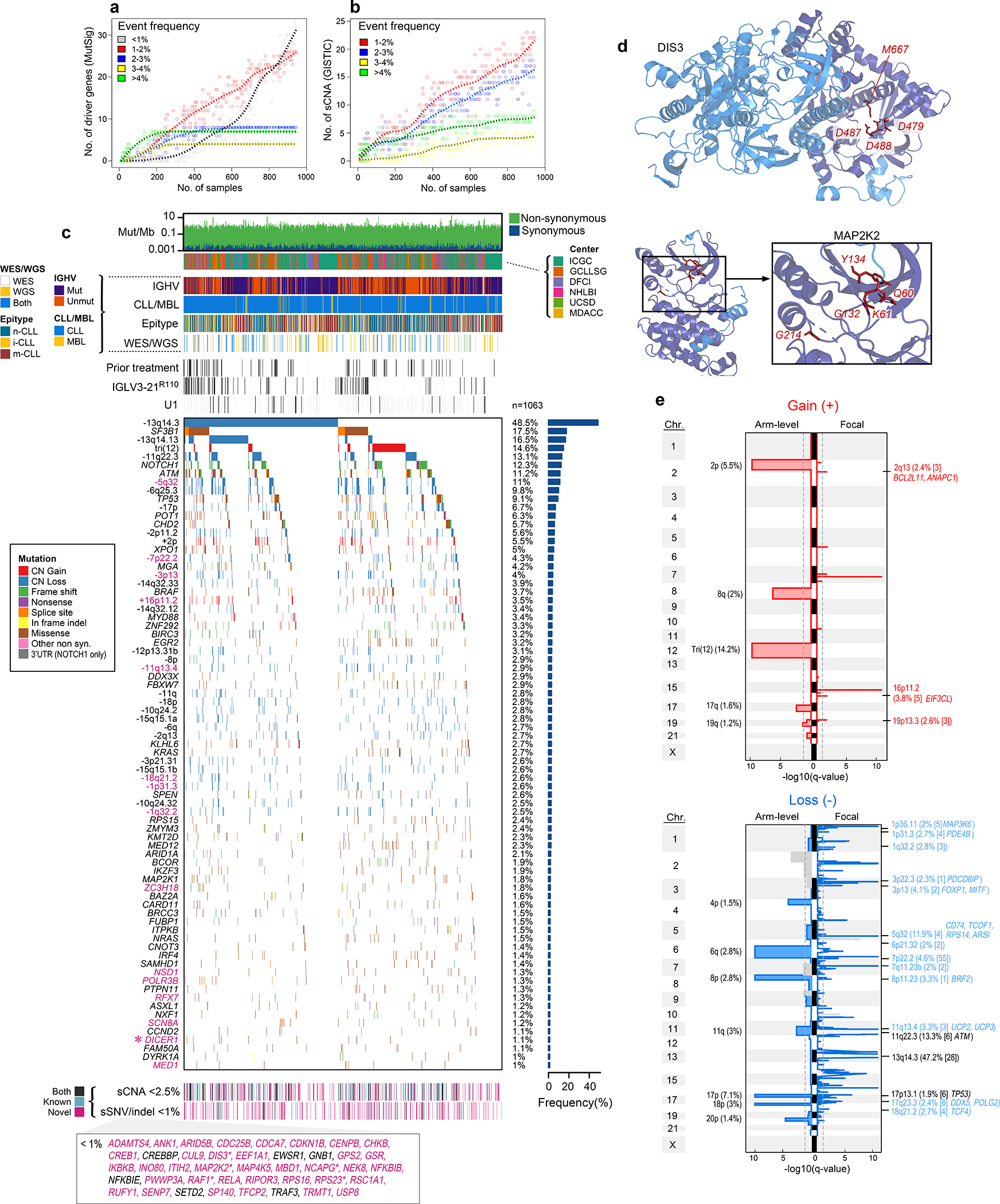
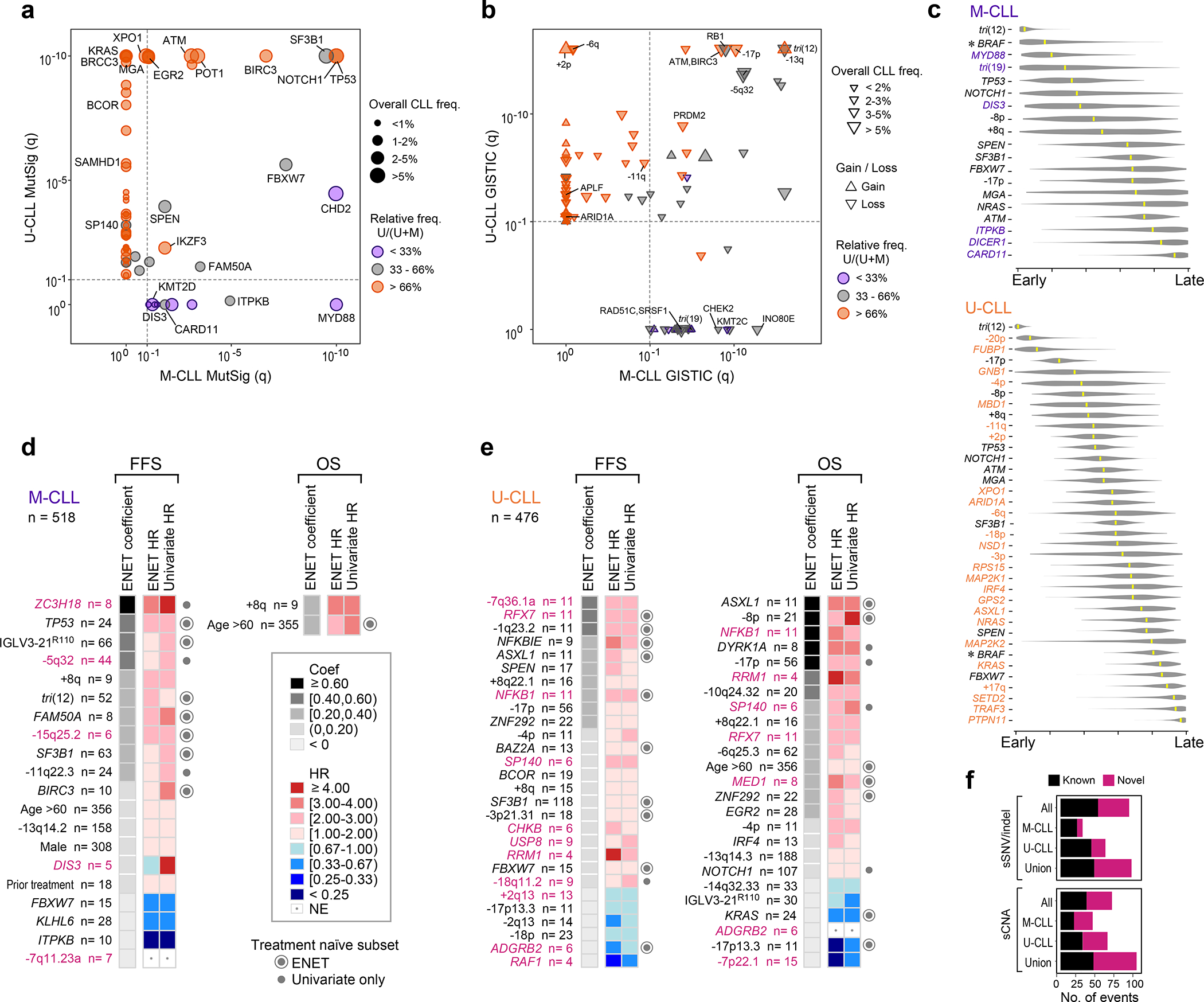
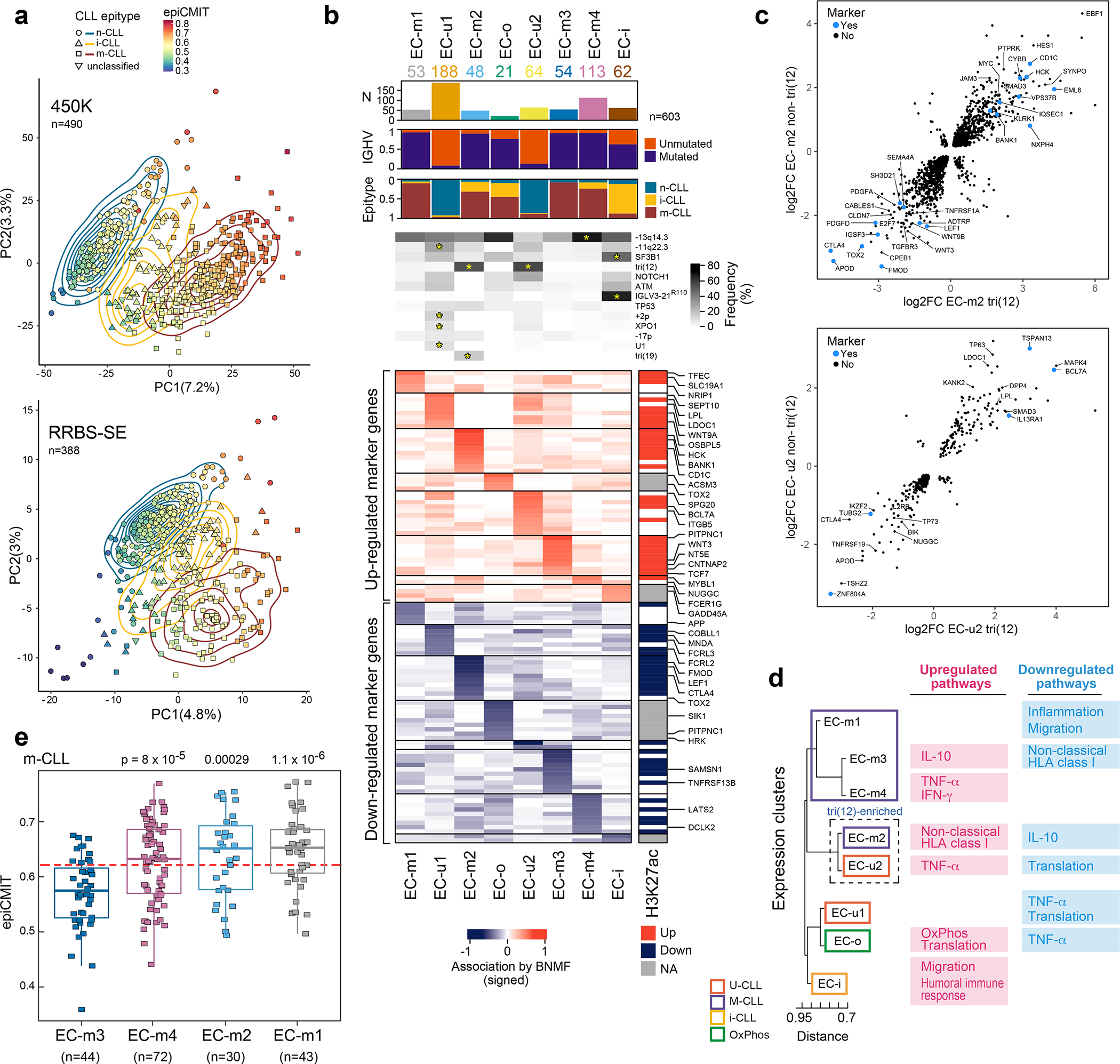
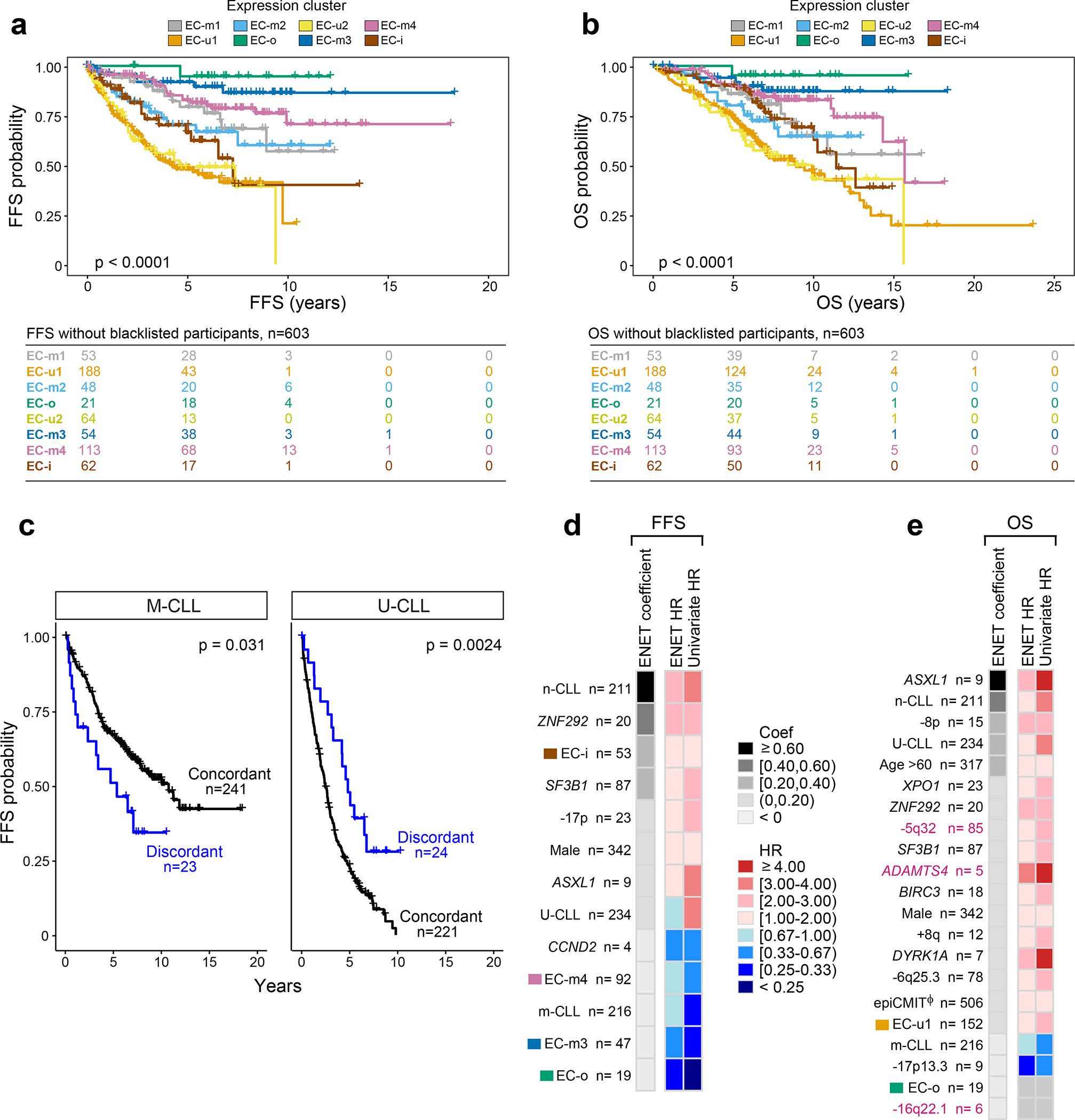
Recent advances in cancer characterization have consistently revealed marked heterogeneity, impeding the completion of integrated molecular and clinical maps for each malignancy. Here, we focus on chronic lymphocytic leukemia (CLL), a B cell neoplasm with variable natural history that is conventionally categorized into two subtypes distinguished by extent of somatic mutations in the heavy-chain variable region of immunoglobulin genes (IGHV). To build the 'CLL map,' we integrated genomic, transcriptomic and epigenomic data from 1,148 patients. We identified 202 candidate genetic drivers of CLL (109 new) and refined the characterization of IGHV subtypes, which revealed distinct genomic landscapes and leukemogenic trajectories. Discovery of new gene expression subtypes further subcategorized this neoplasm and proved to be independent prognostic factors. Clinical outcomes were associated with a combination of genetic, epigenetic and gene expression features, further advancing our prognostic paradigm. Overall, this work reveals fresh insights into CLL oncogenesis and prognostication.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
COMPETING INTEREST DECLARATION
The authors declare the following conflicts related to the CLLmap project: C.J.W. receives research support from Pharmacyclics. E.C. has been a consultant for Illumina. G.G. receives research funds from IBM and Pharmacyclics; and is an inventor on patent applications related to SignatureAnalyzer-GPU. S.S. reports honoraria for consultancy, advisory board membership, speaker honoraria, research grants and travel support from AbbVie, Amgen, AstraZeneca, Celgene, Gilead, GSK, Hoffmann La-Roche, Janssen, Novartis. C.J.W., G.G., B.A.K., Z.L. and C.K.H. are inventors on a patent “Compositions, panels, and methods for characterizing chronic lymphocytic leukemia” (PCT/US21/45144). The following conflicts are unrelated to the CLLmap project: F.N. has received honoraria from Janssen for speaking at educational activities. E.T. declares research support by Abbvie and Roche; Advisory Boards and Speakers Bureau for Janssen, Abbvie and Roche. A.W. received research funding from Pharmacyclics, Acerta, Merck, Verastem, Genmab, Nurix. J.R.B. has served as a consultant for Abbvie, Acerta/Astra-Zeneca, Beigene, Bristol-Myers Squibb/Juno/Celgene, Catapult, Genentech/Roche, Janssen, MEI Pharma, Morphosys AG, Novartis, Pfizer, Rigel; received research funding from Gilead, Loxo/Lilly, Verastem/SecuraBio, Sun, TG Therapeutics; and served on the data safety monitoring committee for Invectys. J.A.B. received research support from AstraZeneca, BeiGene, Gilead, and Pharmacyclics; travel and speaker honoraria from Janssen. X.S.P. is a cofounder of and holds an equity stake in DREAMgenics. C.J.W. holds equity in BioNTech, Inc.. E.C. has been a consultant for Takeda and NanoString Technologies; has received honoraria from Janssen and Roche for speaking at educational activities; and is an inventor on a Lymphoma and Leukemia Molecular Profiling Project patent “Method for subtyping lymphoma subtypes by means of expression profiling” (PCT/US2014/64161). G.G. is an inventor on patent applications related to MSMuTect, MSMutSig, MSIDetect, and POLYSOLVER; and is a founder and consultant of and holds privately held equity in Scorpion Therapeutics. The other authors have no competing interests to declare.
Figures
References
-
- Puente XS et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 526, 519 (2015). - PubMed
METHODS-ONLY REFERENCES
-
- Stilgenbauer S et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood 123, 3247–3254 (2014). - PubMed
-
- Stilgenbauer S et al. Alemtuzumab combined with dexamethasone, followed by alemtuzumab maintenance or Allo-SCT in ‘ultra high-risk’ CLL: Final results from the CLL2O phase II study. Blood 124, 1991–1991 (2014).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
