

ImputAccur: fast and user-friendly calculation of genotype-imputation accuracy-measures
- PMID: 35927623
- PMCID: PMC9351229
- DOI: 10.1186/s12859-022-04863-z
ImputAccur: fast and user-friendly calculation of genotype-imputation accuracy-measures
Abstract
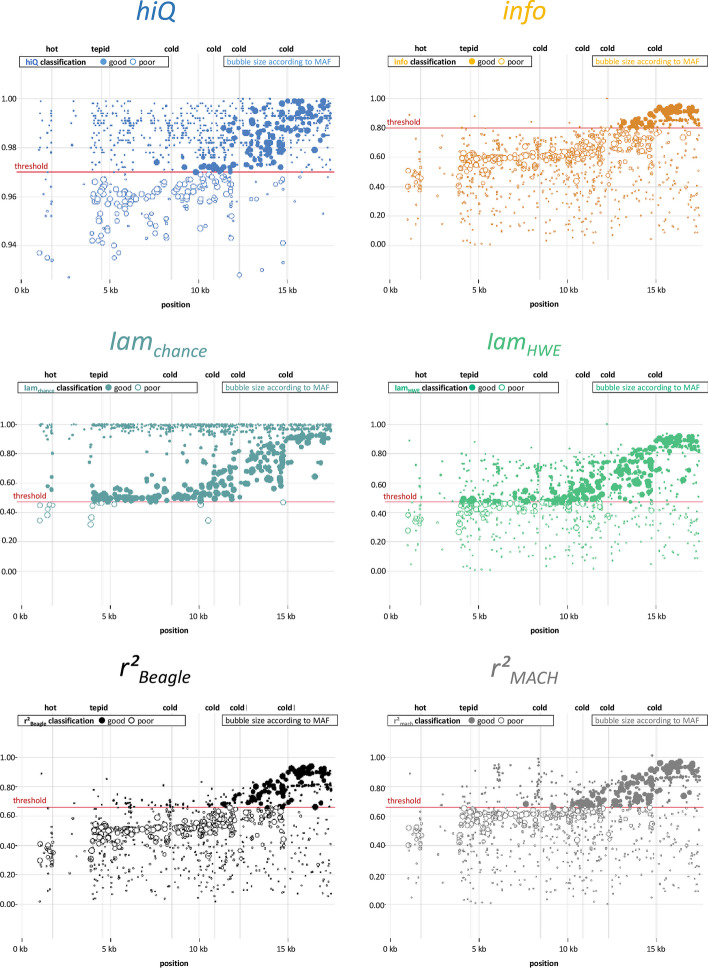
Background: ImputAccur is a software tool to measure genotype-imputation accuracy. Imputation of untyped markers is a standard approach in genome-wide association studies to close the gap between directly genotyped and other known DNA variants. However, high accuracy for imputed genotypes is fundamental. Several accuracy measures have been proposed, but unfortunately, they are implemented on different platforms, which is impractical.
Results: With ImputAccur, the accuracy measures info, Iam-hiQ and r2-based indices can be derived from standard output files of imputation software. Sample/probe and marker filtering is possible. This allows e.g. accurate marker filtering ahead of data analysis.
Conclusions: The source code (Python version 3.9.4), a standalone executive file, and example data for ImputAccur are freely available at https://gitlab.gwdg.de/kolja.thormann1/imputationquality.git .
Keywords: Accuracy; GWAS; Imputation; Marker selection; Quality control; SNP.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Iam hiQ-a novel pair of accuracy indices for imputed genotypes.BMC Bioinformatics. 2022 Jan 24;23(1):50. doi: 10.1186/s12859-022-04568-3. BMC Bioinformatics. 2022. PMID: 35073846 Free PMC article.
-
Accuracy of genome-wide imputation of untyped markers and impacts on statistical power for association studies.BMC Genet. 2009 Jun 16;10:27. doi: 10.1186/1471-2156-10-27. BMC Genet. 2009. PMID: 19531258 Free PMC article.
-
Empirical versus estimated accuracy of imputation: optimising filtering thresholds for sequence imputation.Genet Sel Evol. 2024 Nov 15;56(1):72. doi: 10.1186/s12711-024-00942-2. Genet Sel Evol. 2024. PMID: 39548370 Free PMC article.
-
Accurate Imputation of Untyped Variants from Deep Sequencing Data.Methods Mol Biol. 2021;2243:271-281. doi: 10.1007/978-1-0716-1103-6_13. Methods Mol Biol. 2021. PMID: 33606262 Review.
-
Evaluation of measures of correctness of genotype imputation in the context of genomic prediction: a review of livestock applications.Animal. 2014 Nov;8(11):1743-53. doi: 10.1017/S1751731114001803. Epub 2014 Jul 21. Animal. 2014. PMID: 25045914 Review.
Cited by
-
Aggregating single nucleotide polymorphisms improves filtering for false-positive associations postimputation.G3 (Bethesda). 2025 May 8;15(5):jkaf043. doi: 10.1093/g3journal/jkaf043. G3 (Bethesda). 2025. PMID: 40053832 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
