

Methylome decoding of RdDM-mediated reprogramming effects in the Arabidopsis MSH1 system
- PMID: 35927734
- PMCID: PMC9351182
- DOI: 10.1186/s13059-022-02731-w
Methylome decoding of RdDM-mediated reprogramming effects in the Arabidopsis MSH1 system
Abstract
Background: Plants undergo programmed chromatin changes in response to environment, influencing heritable phenotypic plasticity. The RNA-directed DNA methylation (RdDM) pathway is an essential component of this reprogramming process. The relationship of epigenomic changes to gene networks on a genome-wide basis has been elusive, particularly for intragenic DNA methylation repatterning.
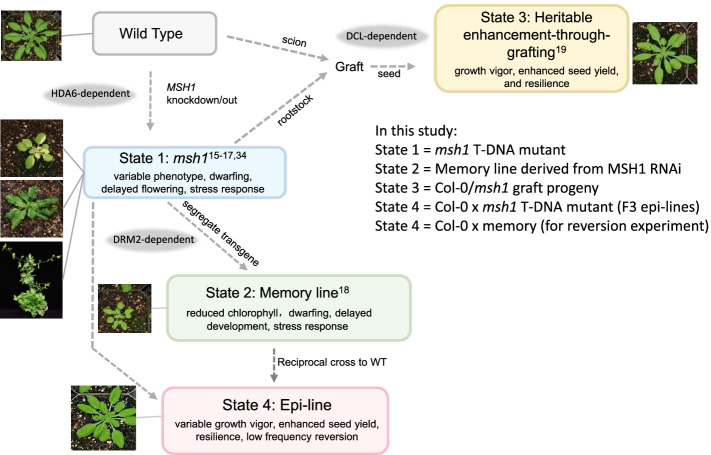
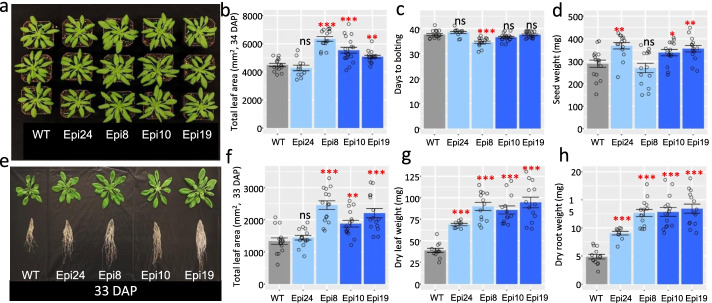
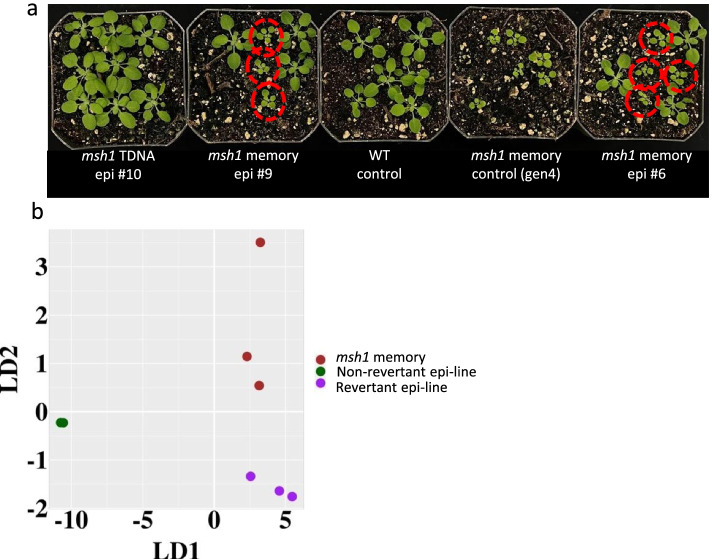
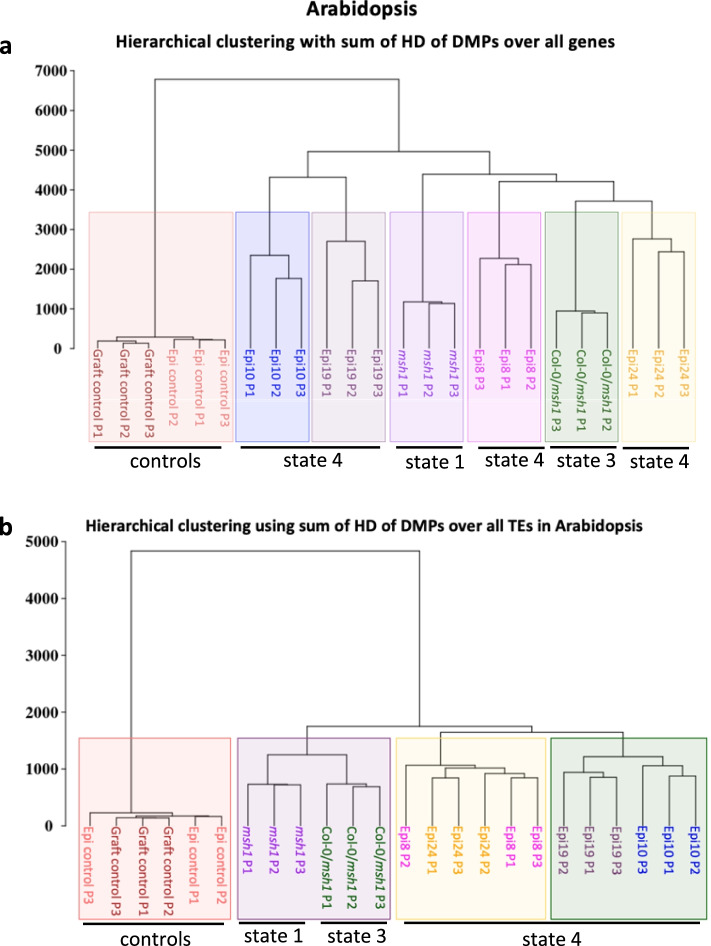
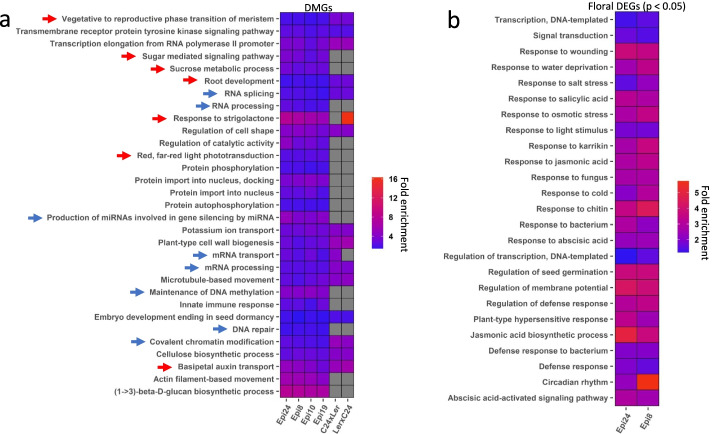
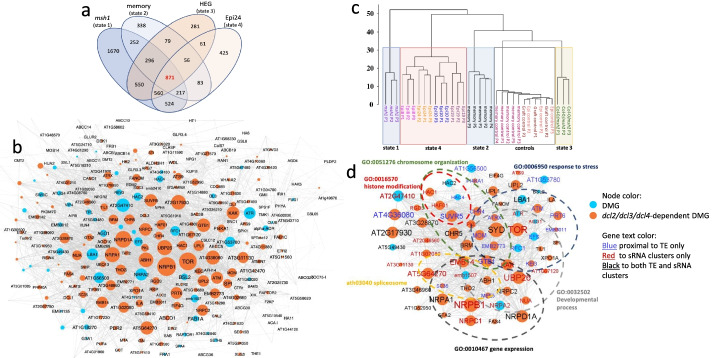
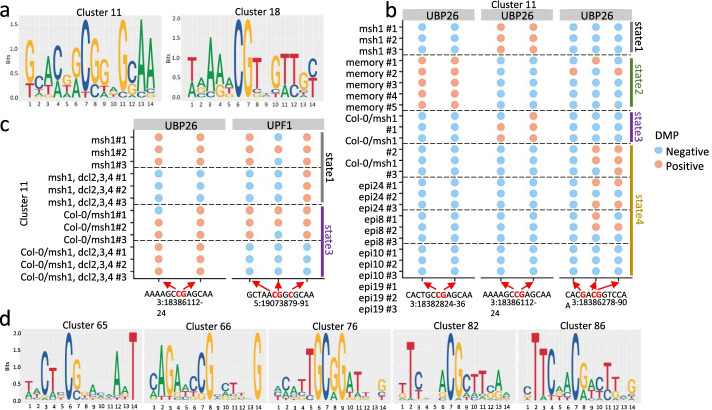
Results: Epigenomic reprogramming is tractable to detailed study and cross-species modeling in the MSH1 system, where perturbation of the plant-specific gene MSH1 triggers at least four distinct nongenetic states to impact plant stress response and growth vigor. Within this system, we have defined RdDM target loci toward decoding phenotype-relevant methylome data. We analyze intragenic methylome repatterning associated with phenotype transitions, identifying state-specific cytosine methylation changes in pivotal growth-versus-stress, chromatin remodeling, and RNA spliceosome gene networks that encompass 871 genes. Over 77% of these genes, and 81% of their central network hubs, are functionally confirmed as RdDM targets based on analysis of mutant datasets and sRNA cluster associations. These dcl2/dcl3/dcl4-sensitive gene methylation sites, many present as singular cytosines, reside within identifiable sequence motifs. These data reflect intragenic methylation repatterning that is targeted and amenable to prediction.
Conclusions: A prevailing assumption that biologically relevant DNA methylation variation occurs predominantly in density-defined differentially methylated regions overlooks behavioral features of intragenic, single-site cytosine methylation variation. RdDM-dependent methylation changes within identifiable sequence motifs reveal gene hubs within networks discriminating stress response and growth vigor epigenetic phenotypes. This study uncovers components of a methylome "code" for de novo intragenic methylation repatterning during plant phenotype transitions.
Keywords: DNA methylation; Epigenetic; Phenotypic plasticity; Stress response; Vigor; sRNA.
© 2022. The Author(s).
Conflict of interest statement
S. Mackenzie is a co-founder for EpiCrop Technologies Inc, a small start-up company that investigates the
Figures

Similar articles
-
MSH1-induced heritable enhanced growth vigor through grafting is associated with the RdDM pathway in plants.Nat Commun. 2020 Oct 22;11(1):5343. doi: 10.1038/s41467-020-19140-x. Nat Commun. 2020. PMID: 33093443 Free PMC article.
-
Segregation of an MSH1 RNAi transgene produces heritable non-genetic memory in association with methylome reprogramming.Nat Commun. 2020 May 5;11(1):2214. doi: 10.1038/s41467-020-16036-8. Nat Commun. 2020. PMID: 32371941 Free PMC article.
-
Full-length autonomous transposable elements are preferentially targeted by expression-dependent forms of RNA-directed DNA methylation.Genome Biol. 2016 Aug 9;17(1):170. doi: 10.1186/s13059-016-1032-y. Genome Biol. 2016. PMID: 27506905 Free PMC article.
-
Epigenomics in stress tolerance of plants under the climate change.Mol Biol Rep. 2023 Jul;50(7):6201-6216. doi: 10.1007/s11033-023-08539-6. Epub 2023 Jun 9. Mol Biol Rep. 2023. PMID: 37294468 Review.
-
Advances of RNA polymerase IV in controlling DNA methylation and development in plants.Yi Chuan. 2022 Jul 20;44(7):567-580. doi: 10.16288/j.yczz.22-063. Yi Chuan. 2022. PMID: 35858769 Review.
Cited by
-
Flipping the switch on some of the slowest mutating genomes: Direct measurements of plant mitochondrial and plastid mutation rates in msh1 mutants.bioRxiv [Preprint]. 2025 Jan 11:2025.01.08.631957. doi: 10.1101/2025.01.08.631957. bioRxiv. 2025. Update in: PLoS Genet. 2025 Jun 30;21(6):e1011764. doi: 10.1371/journal.pgen.1011764. PMID: 39829752 Free PMC article. Updated. Preprint.
-
Expansion of the MutS gene family in plants.Plant Cell. 2025 Jul 1;37(7):koae277. doi: 10.1093/plcell/koae277. Plant Cell. 2025. PMID: 39692564 Free PMC article.
-
Phenotypic plasticity for improved light harvesting, in tandem with methylome repatterning in reef-building corals.Mol Ecol. 2024 Feb;33(4):e17246. doi: 10.1111/mec.17246. Epub 2023 Dec 28. Mol Ecol. 2024. PMID: 38153177 Free PMC article.
-
Short-Term High Light Stress Analysis Through Differential Methylation Identifies Root Architecture and Cell Size Responses.Plant Cell Environ. 2025 May;48(5):3269-3280. doi: 10.1111/pce.15325. Epub 2024 Dec 25. Plant Cell Environ. 2025. PMID: 39722567 Free PMC article.
-
Expansion of the MutS Gene Family in Plants.bioRxiv [Preprint]. 2024 Jul 20:2024.07.17.603841. doi: 10.1101/2024.07.17.603841. bioRxiv. 2024. Update in: Plant Cell. 2025 Jul 1;37(7):koae277. doi: 10.1093/plcell/koae277. PMID: 39071318 Free PMC article. Updated. Preprint.
References
-
- Gunn CR, Dennis JV. World Guide to Tropical Drift Seeds and Fruits. NY: Quadrangle/New York Times Book Co; 1976.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
