

Construction of a cross-species cell landscape at single-cell level
- PMID: 35929025
- PMCID: PMC9881150
- DOI: 10.1093/nar/gkac633
Construction of a cross-species cell landscape at single-cell level
Abstract
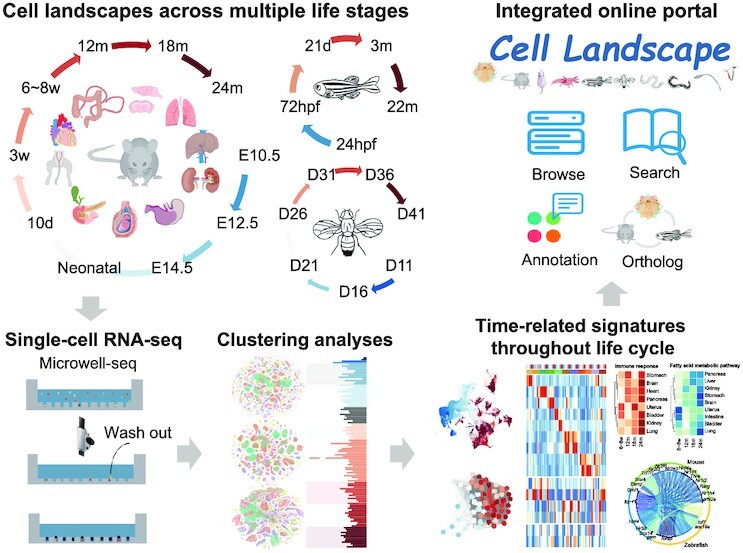
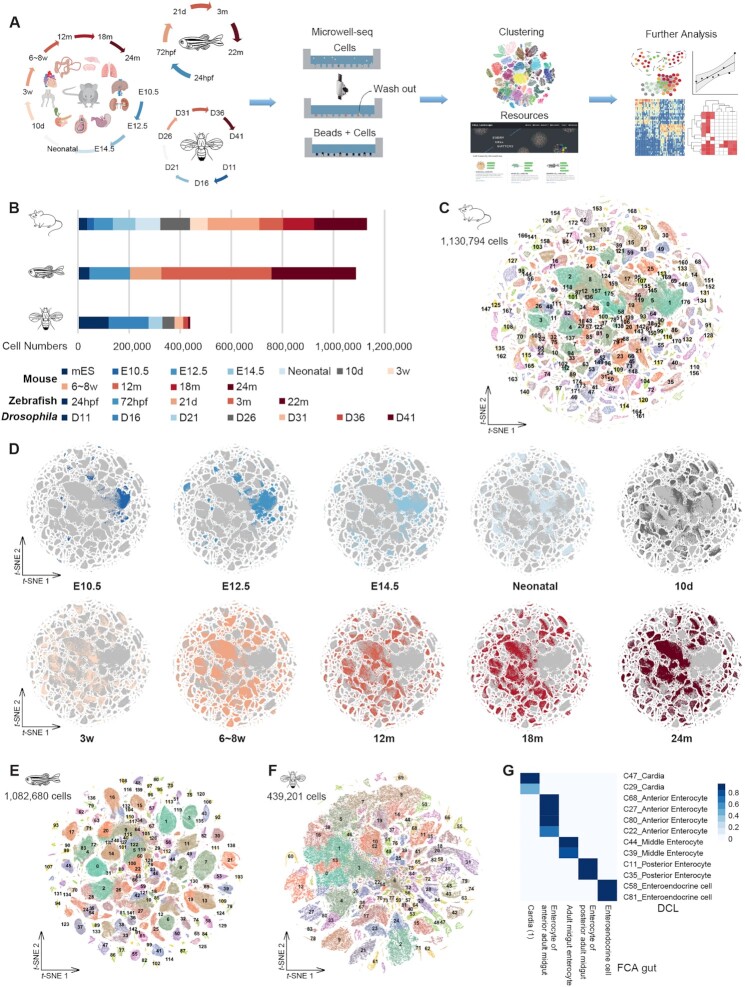
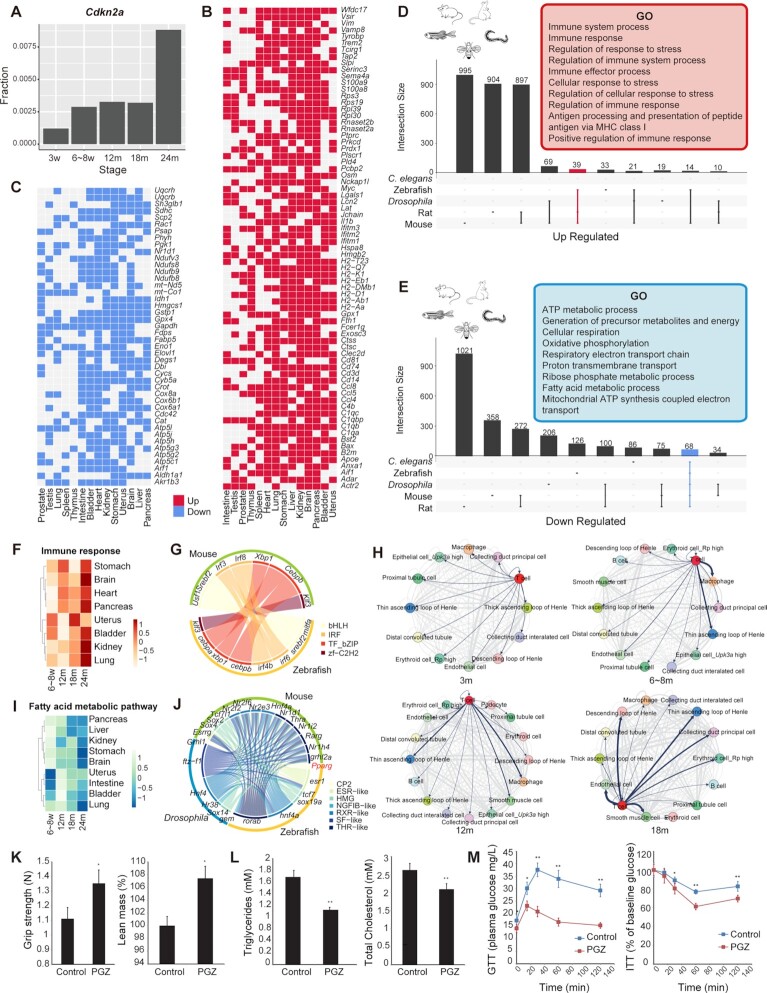
Individual cells are basic units of life. Despite extensive efforts to characterize the cellular heterogeneity of different organisms, cross-species comparisons of landscape dynamics have not been achieved. Here, we applied single-cell RNA sequencing (scRNA-seq) to map organism-level cell landscapes at multiple life stages for mice, zebrafish and Drosophila. By integrating the comprehensive dataset of > 2.6 million single cells, we constructed a cross-species cell landscape and identified signatures and common pathways that changed throughout the life span. We identified structural inflammation and mitochondrial dysfunction as the most common hallmarks of organism aging, and found that pharmacological activation of mitochondrial metabolism alleviated aging phenotypes in mice. The cross-species cell landscape with other published datasets were stored in an integrated online portal-Cell Landscape. Our work provides a valuable resource for studying lineage development, maturation and aging.
Plain language summary
How many cell types are there in nature? How do they change during the life cycle? These are two fundamental questions that researchers have been trying to understand in the area of biology. In this study, single-cell mRNA sequencing data were used to profile over 2.6 million individual cells from mice, zebrafish and Drosophila at different life stages, 1.3 million of which were newly collected. The comprehensive datasets allow investigators to construct a cross-species cell landscape that helps to reveal the conservation and diversity of cell taxonomies at genetic and regulatory levels. The resources in this study are assembled into a publicly available website at http://bis.zju.edu.cn/cellatlas/.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures


References
-
- Hashimshony T., Wagner F., Sher N., Yanai I.. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Rep. 2012; 2:666–673. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
