

Transthyretin cardiac amyloidosis
- PMID: 35929637
- PMCID: PMC9897687
- DOI: 10.1093/cvr/cvac119
Transthyretin cardiac amyloidosis
Abstract
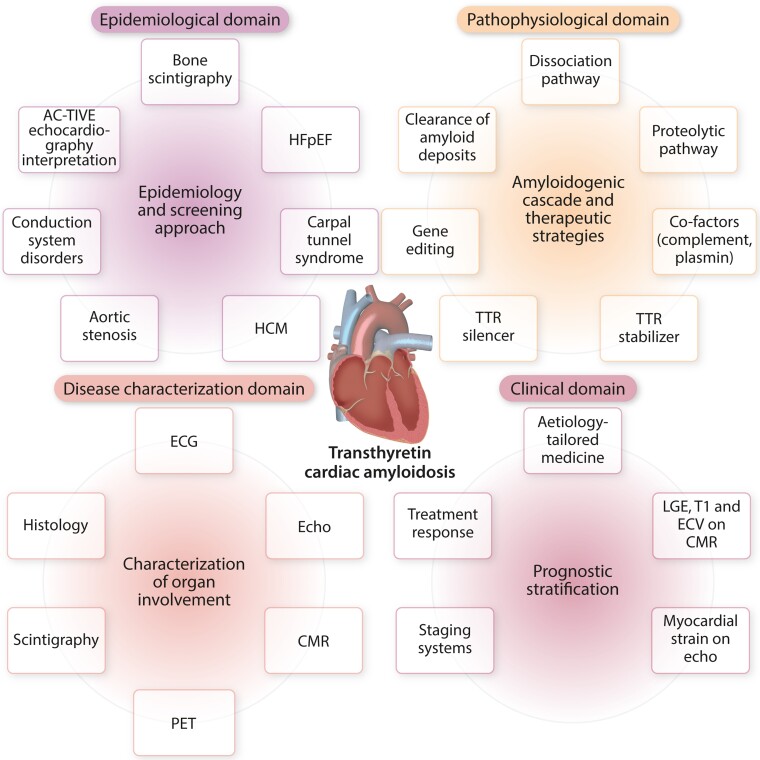
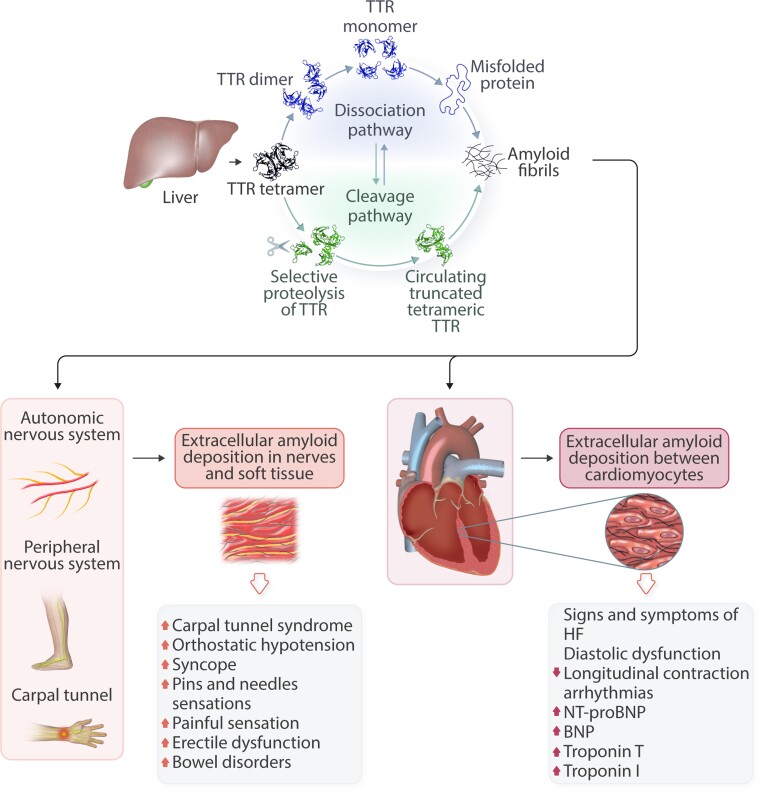
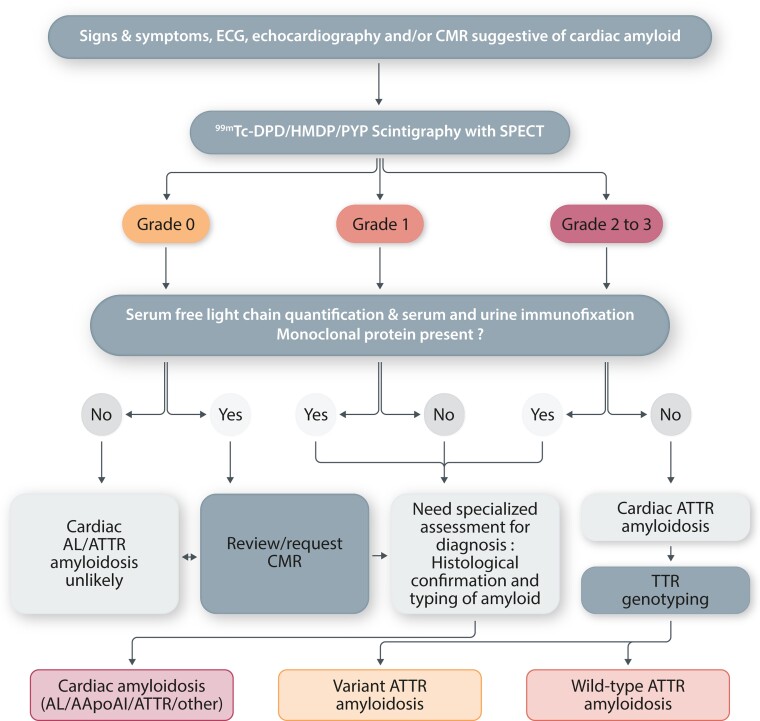
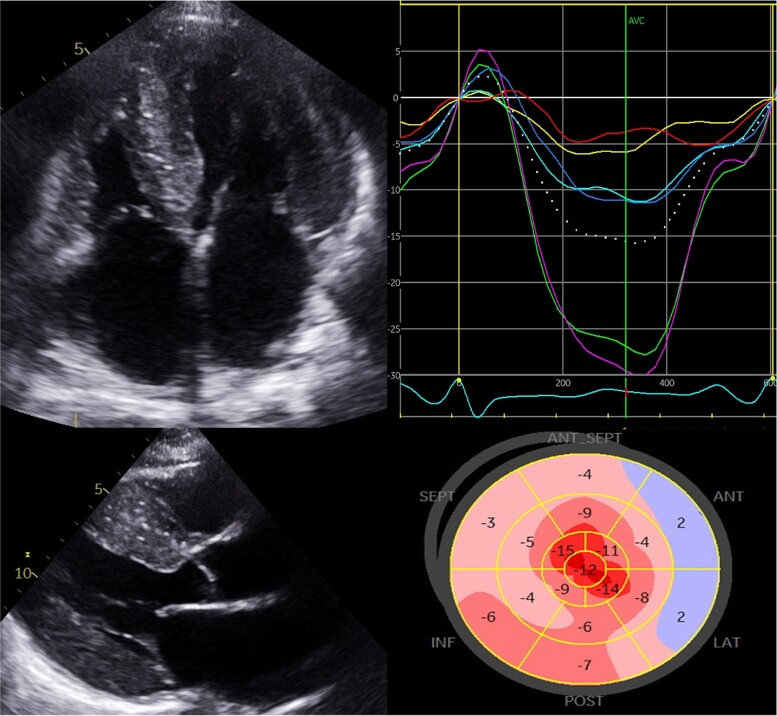
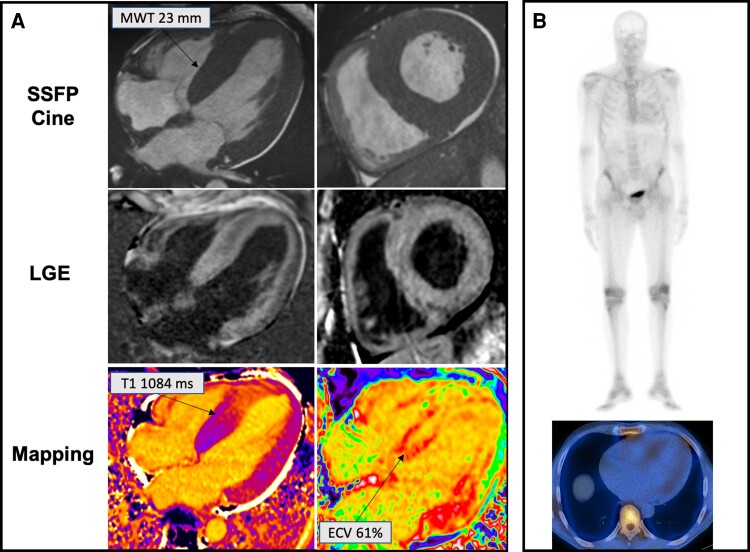
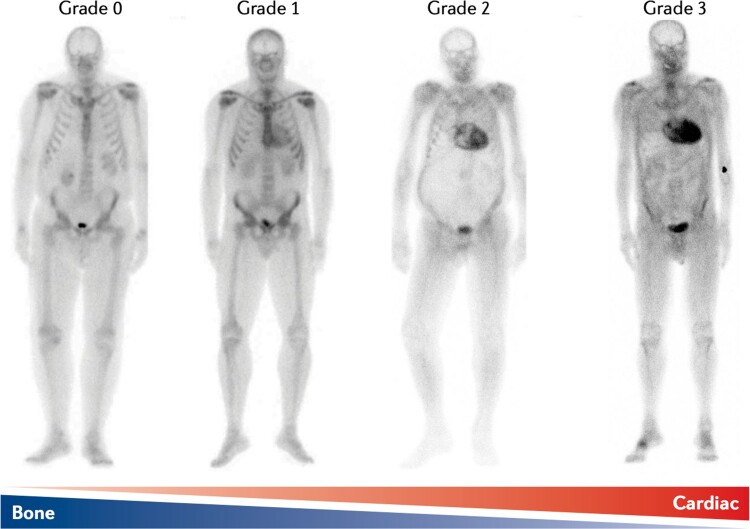
Transthyretin cardiac amyloidosis (ATTR-CA) is an increasingly recognized cause of heart failure (HF) and mortality worldwide. Advances in non-invasive diagnosis, coupled with the development of effective treatments, have shifted ATTR-CA from a rare and untreatable disease to a relatively prevalent condition that clinicians should consider on a daily basis. Amyloid fibril formation results from age-related failure of homoeostatic mechanisms in wild-type ATTR (ATTRwt) amyloidosis (non-hereditary form) or destabilizing mutations in variant ATTR (ATTRv) amyloidosis (hereditary form). Longitudinal large-scale studies in the United States suggest an incidence of cardiac amyloidosis in the contemporary era of 17 per 100 000, which has increased from a previous estimate of 0.5 per 100 000, which was almost certainly due to misdiagnosis and underestimated. The presence and degree of cardiac involvement is the leading cause of mortality both in ATTRwt and ATTRv amyloidosis, and can be identified in up to 15% of patients hospitalized for HF with preserved ejection fraction. Associated features, such as carpal tunnel syndrome, can preceed by several years the development of symptomatic HF and may serve as early disease markers. Echocardiography and cardiac magnetic resonance raise suspicion of disease and might offer markers of treatment response at a myocardial level, such as extracellular volume quantification. Radionuclide scintigraphy with 'bone' tracers coupled with biochemical tests may differentiate ATTR from light chain amyloidosis. Therapies able to slow or halt ATTR-CA progression and increase survival are now available. In this evolving scenario, early disease recognition is paramount to derive the greatest benefit from treatment.
Keywords: Cardiac magnetic resonance; Cardiac scintigraphy with bone tracers; Prognostic stratification; TTR; Therapy; Transthyretin cardiac amyloidosis.
© The Author(s) 2022. Published by Oxford University Press on behalf of the European Society of Cardiology.
Conflict of interest statement
Conflict of interest: J.D.G: expert advisor for Alnylam, Ionis, Astra-Zenica, Eidos, Intellia and Pfizer. Other authors report no conflict of interest to declare.
Figures
References
-
- Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet 2016;387:2641–2654. - PubMed
-
- Martinez-Naharro A, Baksi AJ, Hawkins PN, Fontana M. Diagnostic imaging of cardiac amyloidosis. Nat Rev Cardiol 2020;17:413–426. - PubMed
-
- Lavatelli F, Vrana JA. Proteomic typing of amyloid deposits in systemic amyloidoses. Amyloid 2011;18:177–182. - PubMed
-
- Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, Judge DP, Lenihan DJ, Gottlieb SS, Shah SJ, Steidley DE, Ventura H, Murali S, Silver MA, Jacoby D, Fedson S, Hummel SL, Kristen A V, Damy T, Planté-Bordeneuve V, Coelho T, Mundayat R, Suhr OB, Waddington Cruz M, Rapezzi C. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (transthyretin amyloid outcome survey). J Am Coll Cardiol 2016;68:161–172. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
