

The Congenital and Acquired Mechanisms Implicated in the Etiology of Central Precocious Puberty
- PMID: 35930274
- PMCID: PMC9985412
- DOI: 10.1210/endrev/bnac020
The Congenital and Acquired Mechanisms Implicated in the Etiology of Central Precocious Puberty
Erratum in
-
Correction to: "The Congenital and Acquired Mechanisms Implicated in the Etiology of Central Precocious Puberty".Endocr Rev. 2023 Mar 4;44(2):355. doi: 10.1210/endrev/bnac036. Endocr Rev. 2023. PMID: 36656052 Free PMC article. No abstract available.
Abstract
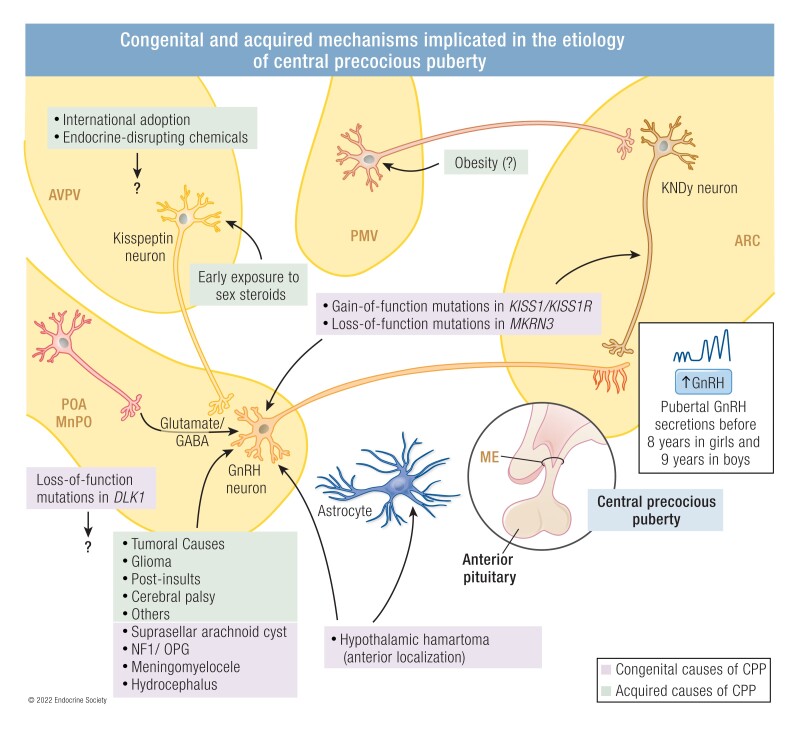
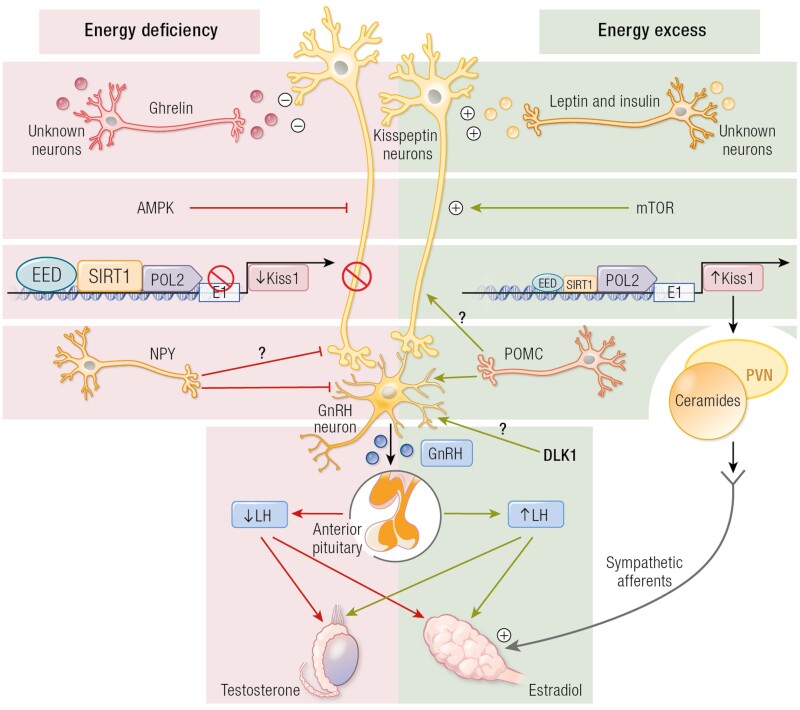
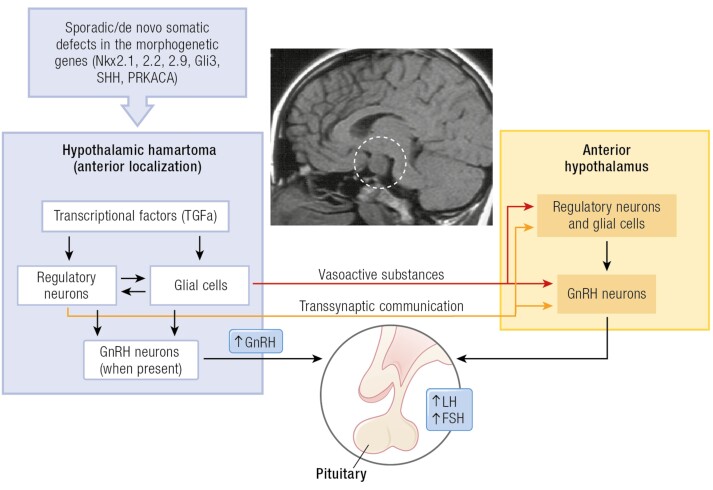
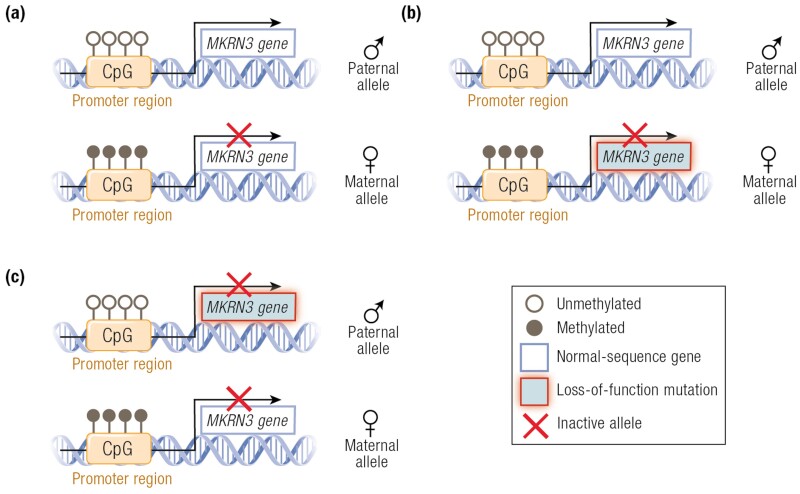
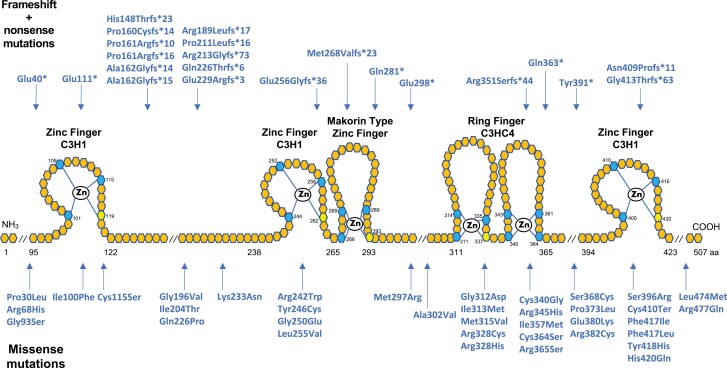
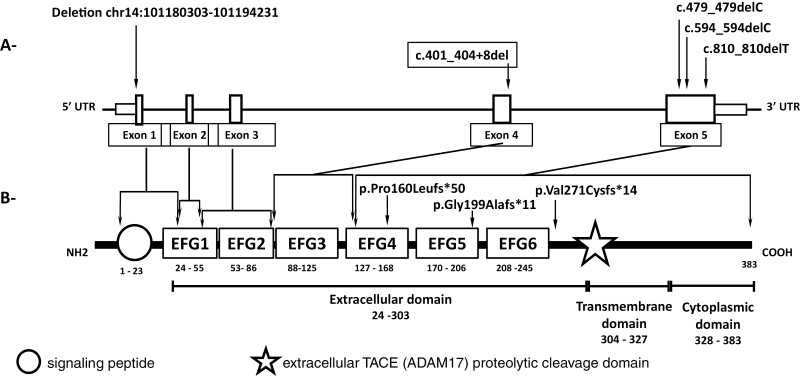
The etiology of central precocious puberty (CPP) is multiple and heterogeneous, including congenital and acquired causes that can be associated with structural or functional brain alterations. All causes of CPP culminate in the premature pulsatile secretion of hypothalamic GnRH and, consequently, in the premature reactivation of hypothalamic-pituitary-gonadal axis. The activation of excitatory factors or suppression of inhibitory factors during childhood represent the 2 major mechanisms of CPP, revealing a delicate balance of these opposing neuronal pathways. Hypothalamic hamartoma (HH) is the most well-known congenital cause of CPP with central nervous system abnormalities. Several mechanisms by which hamartoma causes CPP have been proposed, including an anatomical connection to the anterior hypothalamus, autonomous neuroendocrine activity in GnRH neurons, trophic factors secreted by HH, and mechanical pressure applied to the hypothalamus. The importance of genetic and/or epigenetic factors in the underlying mechanisms of CPP has grown significantly in the last decade, as demonstrated by the evidence of genetic abnormalities in hypothalamic structural lesions (eg, hamartomas, gliomas), syndromic disorders associated with CPP (Temple, Prader-Willi, Silver-Russell, and Rett syndromes), and isolated CPP from monogenic defects (MKRN3 and DLK1 loss-of-function mutations). Genetic and epigenetic discoveries involving the etiology of CPP have had influence on the diagnosis and familial counseling providing bases for potential prevention of premature sexual development and new treatment targets in the future. Global preventive actions inducing healthy lifestyle habits and less exposure to endocrine-disrupting chemicals during the lifespan are desirable because they are potentially associated with CPP.
Keywords: DLK1; MKRN3; central precocious puberty; endocrine-disrupting chemicals; gonadotropin-releasing hormone; hypothalamic hamartoma; kisspeptins.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev. 2003;24(5):668–693. - PubMed
-
- Latronico AC, Brito VN, Carel JC. Causes, diagnosis, and treatment of central precocious puberty. Lancet Diabetes Endocrinol. 2016;4(3):265–274. - PubMed
-
- Carel JC, Léger J. Clinical practice. Precocious puberty. N Engl J Med. 2008;358(22):2366–2377. - PubMed
-
- Sørensen K, Mouritsen A, Aksglaede L, Hagen CP, Mogensen SS, Juul A. Recent secular trends in pubertal timing: implications for evaluation and diagnosis of precocious puberty. Horm Res Paediatr. 2012;77(3):137–145. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
