

An Immunogenic Model of KRAS-Mutant Lung Cancer Enables Evaluation of Targeted Therapy and Immunotherapy Combinations
- PMID: 35930804
- PMCID: PMC7613674
- DOI: 10.1158/0008-5472.CAN-22-0325
An Immunogenic Model of KRAS-Mutant Lung Cancer Enables Evaluation of Targeted Therapy and Immunotherapy Combinations
Abstract
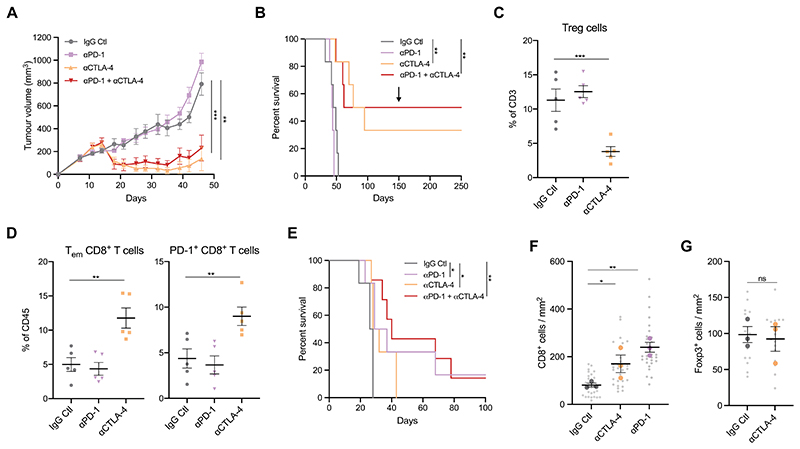
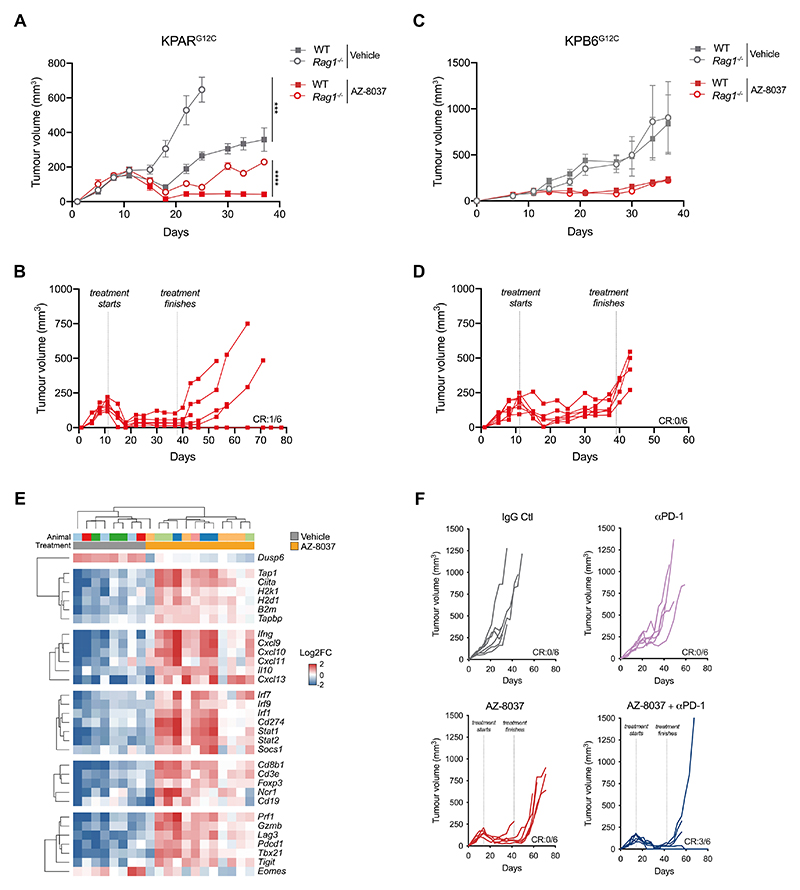
Mutations in oncogenes such as KRAS and EGFR cause a high proportion of lung cancers. Drugs targeting these proteins cause tumor regression but ultimately fail to elicit cures. As a result, there is an intense interest in how to best combine targeted therapies with other treatments, such as immunotherapies. However, preclinical systems for studying the interaction of lung tumors with the host immune system are inadequate, in part due to the low tumor mutational burden in genetically engineered mouse models. Here we set out to develop mouse models of mutant KRAS-driven lung cancer with an elevated tumor mutational burden by expressing the human DNA cytosine deaminase, APOBEC3B, to mimic the mutational signature seen in human lung cancer. This failed to substantially increase clonal tumor mutational burden and autochthonous tumors remained refractory to immunotherapy. However, establishing clonal cell lines from these tumors enabled the generation of an immunogenic syngeneic transplantation model of KRAS-mutant lung adenocarcinoma that was sensitive to immunotherapy. Unexpectedly, antitumor immune responses were not directed against neoantigens but instead targeted derepressed endogenous retroviral antigens. The ability of KRASG12C inhibitors to cause regression of KRASG12C -expressing tumors was markedly potentiated by the adaptive immune system, highlighting the importance of using immunocompetent models for evaluating targeted therapies. Overall, this model provides a unique opportunity for the study of combinations of targeted and immunotherapies in immune-hot lung cancer.
Significance: This study develops a mouse model of immunogenic KRAS-mutant lung cancer to facilitate the investigation of optimal combinations of targeted therapies with immunotherapies.
©2022 American Association for Cancer Research.
Conflict of interest statement
Competing interests: J.D. has acted as a consultant for AstraZeneca, Bayer, Jubilant, Theras, Vividion and Novartis. S.R. is an employee of AstraZeneca. C.S. receives grant support from Archer Dx, AstraZeneca, Boehringer–Ingelheim and Ono Pharmaceutical; has consulted for AstraZeneca, Bicycle Therapeutics, Celgene, Genentech, GRAIL, GSK, Illumina, Medicxi, MSD, Novartis and the Sarah Cannon Research Institute; receives grant support and has consulted for Bristol Myers Squibb, Pfizer and Roche–Ventana; is an advisory board member and is involved in trials sponsored by AstraZeneca; has stock options in Epic Sciences, GRAIL; and has stock options and is a co-founder of Achilles Therapeutics. The other authors declare that they have no potential conflicts of interest.
Figures
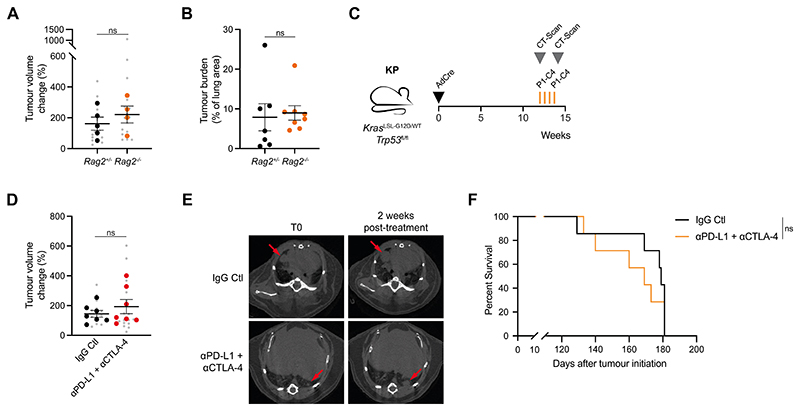
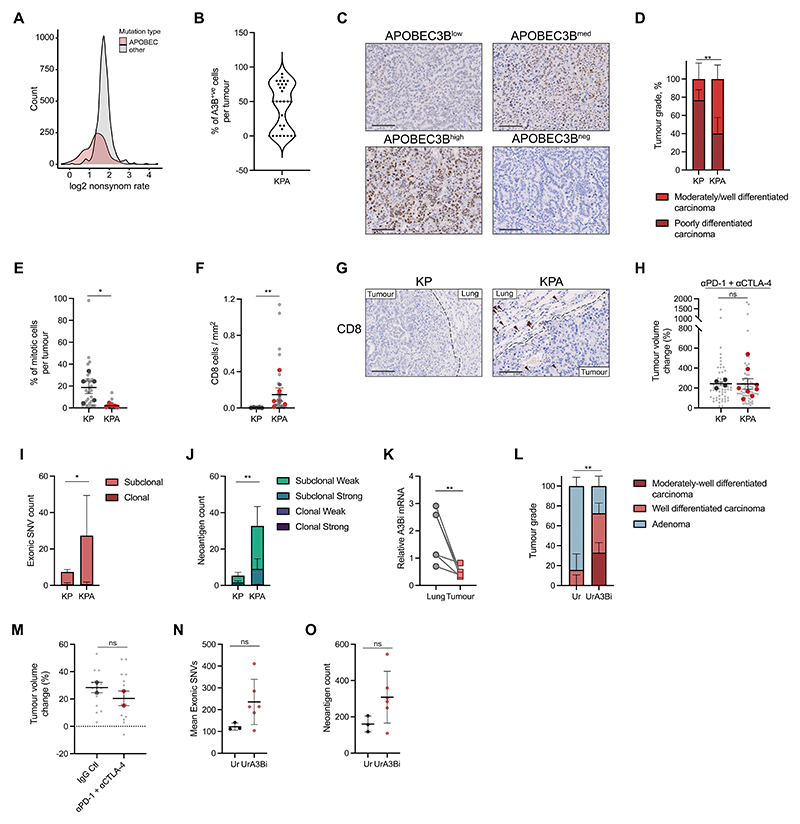
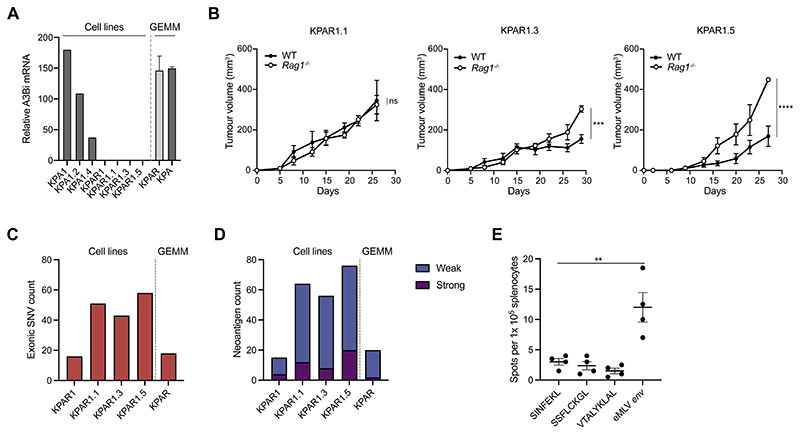
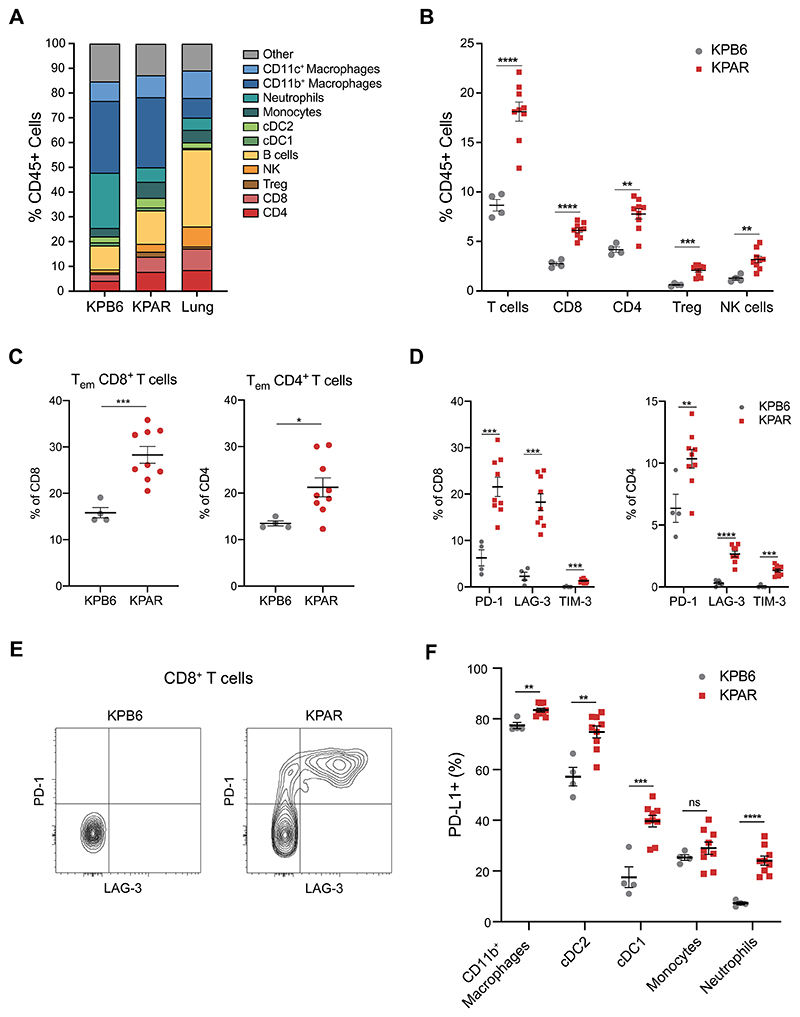
References
-
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. - PubMed
-
- Blumenschein GR, Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC) Ann Oncol. 2015;26:894–901. doi: 10.1093/annonc/mdv072. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
