

A multiomics disease progression signature of low-risk ccRCC
- PMID: 35931808
- PMCID: PMC9356046
- DOI: 10.1038/s41598-022-17755-2
A multiomics disease progression signature of low-risk ccRCC
Abstract
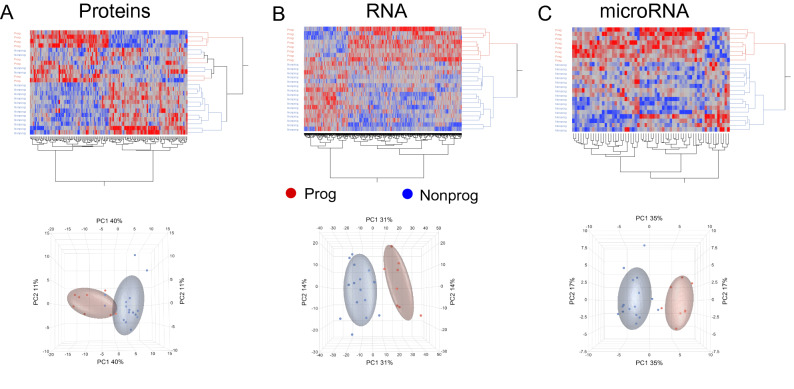
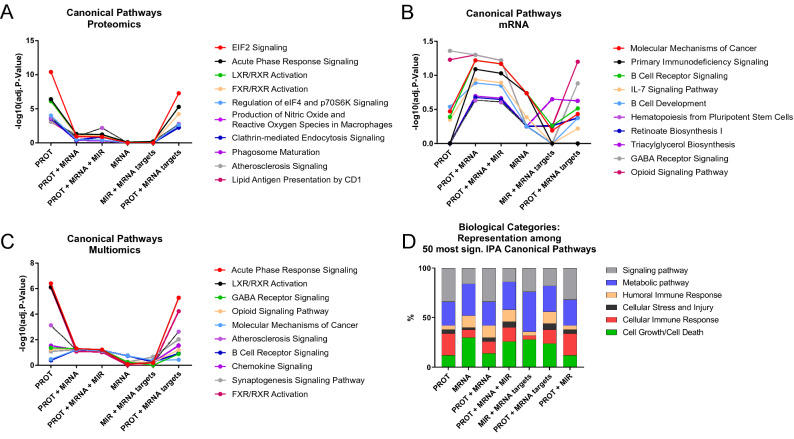
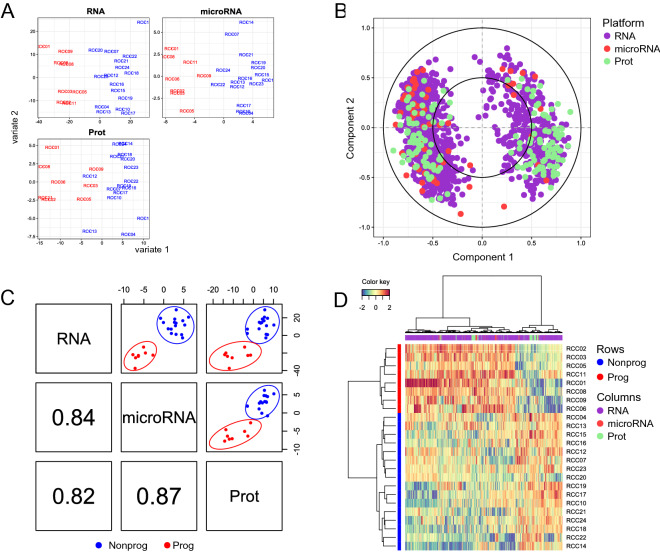
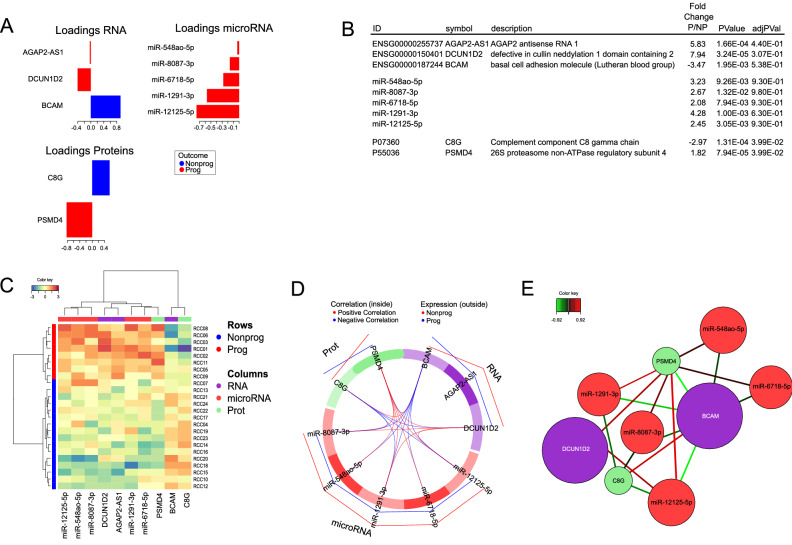
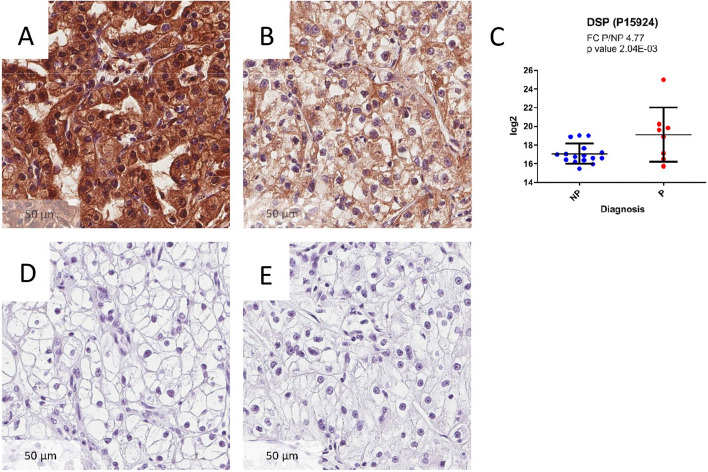
Clear cell renal cell carcinoma (ccRCC) is the most common renal cancer. Identification of ccRCC likely to progress, despite an apparent low risk at the time of surgery, represents a key clinical issue. From a cohort of adult ccRCC patients (n = 443), we selected low-risk tumors progressing within a 5-years average follow-up (progressors: P, n = 8) and non-progressing (NP) tumors (n = 16). Transcriptome sequencing, miRNA sequencing and proteomics were performed on tissues obtained at surgery. We identified 151 proteins, 1167 mRNAs and 63 miRNAs differentially expressed in P compared to NP low-risk tumors. Pathway analysis demonstrated overrepresentation of proteins related to "LXR/RXR and FXR/RXR Activation", "Acute Phase Response Signaling" in NP compared to P samples. Integrating mRNA, miRNA and proteomic data, we developed a 10-component classifier including two proteins, three genes and five miRNAs, effectively differentiating P and NP ccRCC and capturing underlying biological differences, potentially useful to identify "low-risk" patients requiring closer surveillance and treatment adjustments. Key results were validated by immunohistochemistry, qPCR and data from publicly available databases. Our work suggests that LXR, FXR and macrophage activation pathways could be critically involved in the inhibition of the progression of low-risk ccRCC. Furthermore, a 10-component classifier could support an early identification of apparently low-risk ccRCC patients.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Ljungberg B, Albiges L, Abu-Ghanem Y, Bensalah K, Dabestani S, Montes SF, et al. European association of urology guidelines on renal cell carcinoma: The 2019 update. Eur. Urol. 2019;1:1. - PubMed
-
- Voss J, Drake T, Matthews H, Jenkins J, Tang S, Doherty J, et al. Chest computed tomography for staging renal tumours: Validation and simplification of a risk prediction model from a large contemporary retrospective cohort. BJU Int. 2020;1:1. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
