

Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment
- PMID: 35931863
- PMCID: PMC9886402
- DOI: 10.1038/s41588-022-01141-9
Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment
Abstract
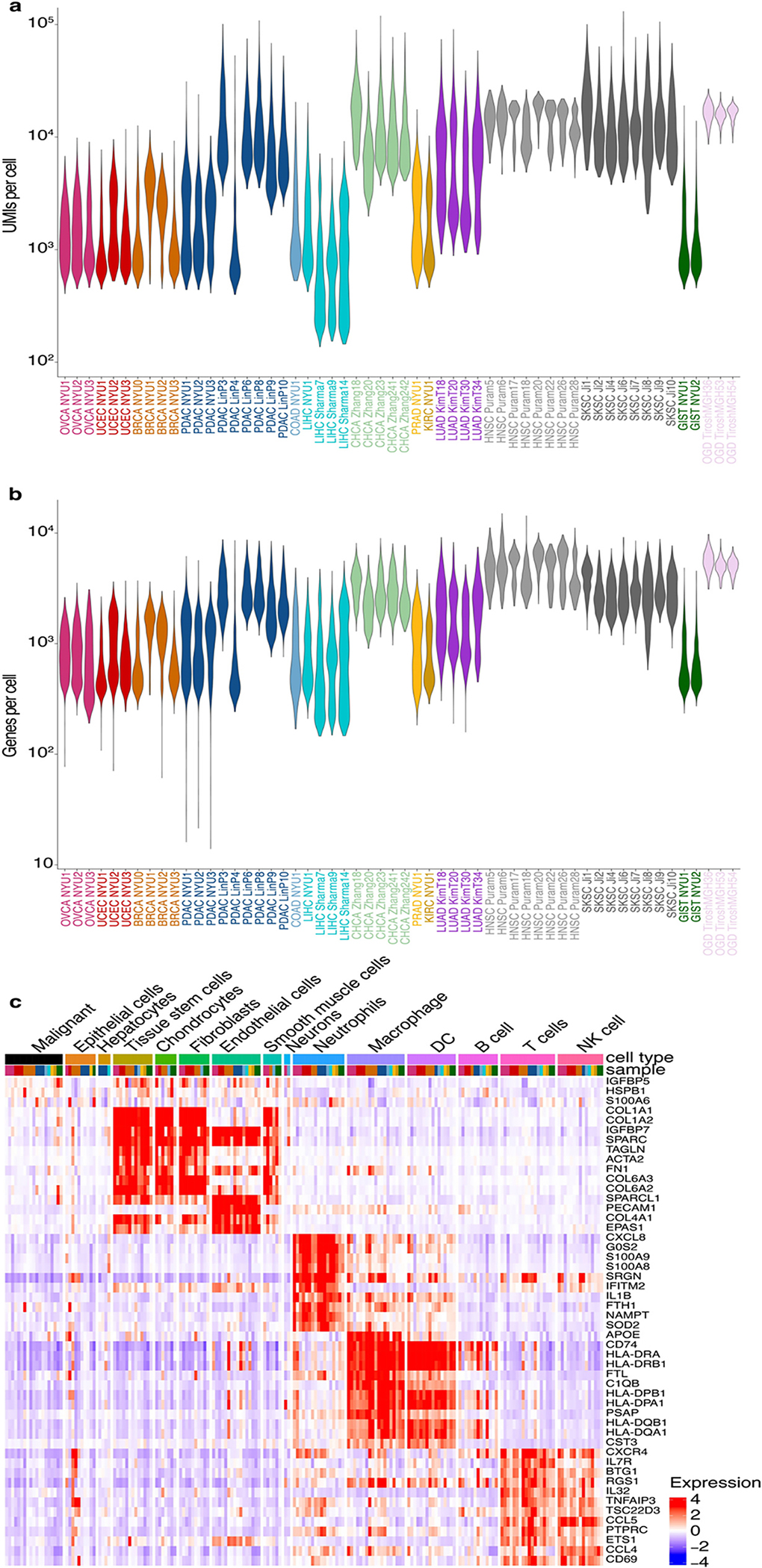
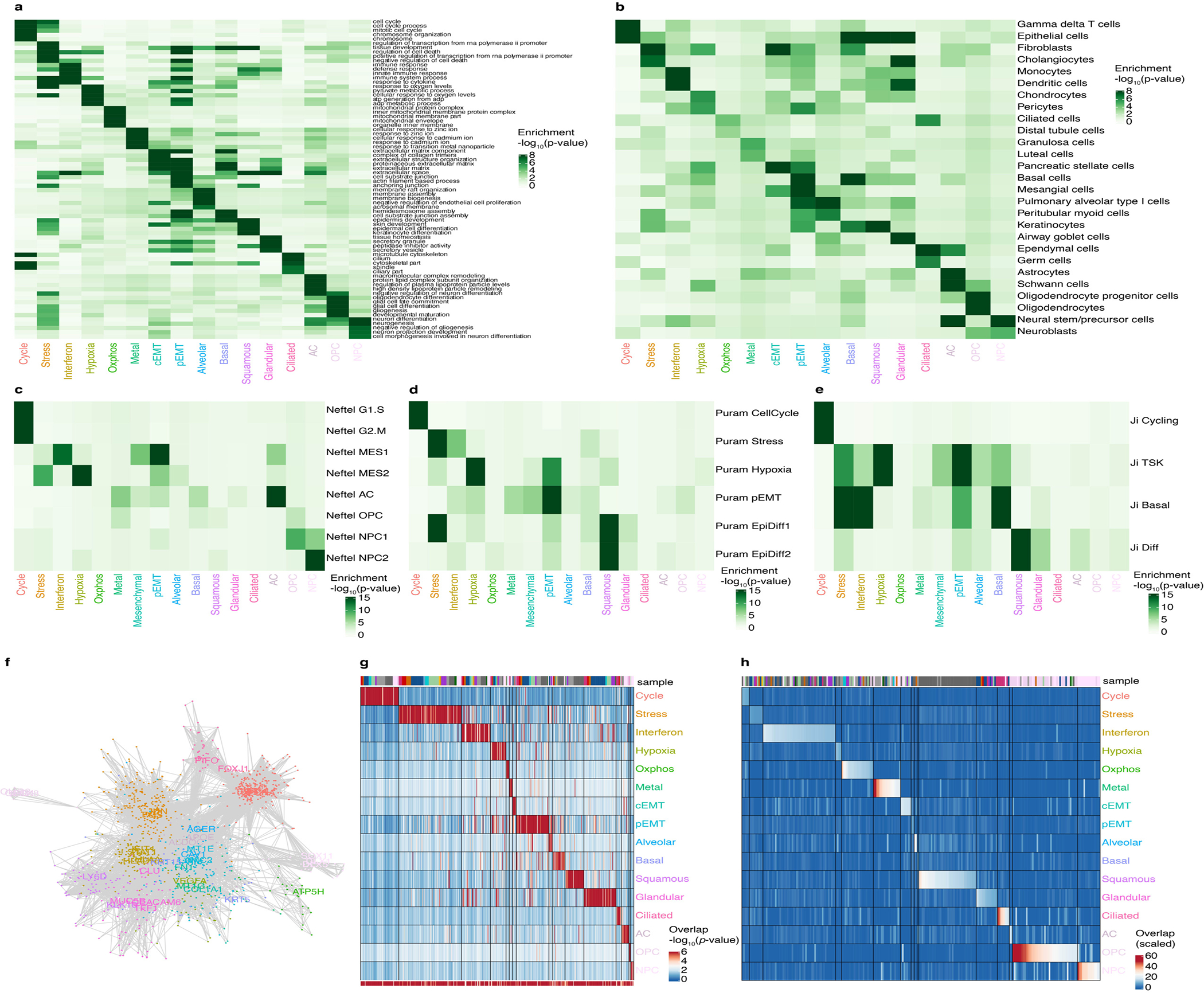
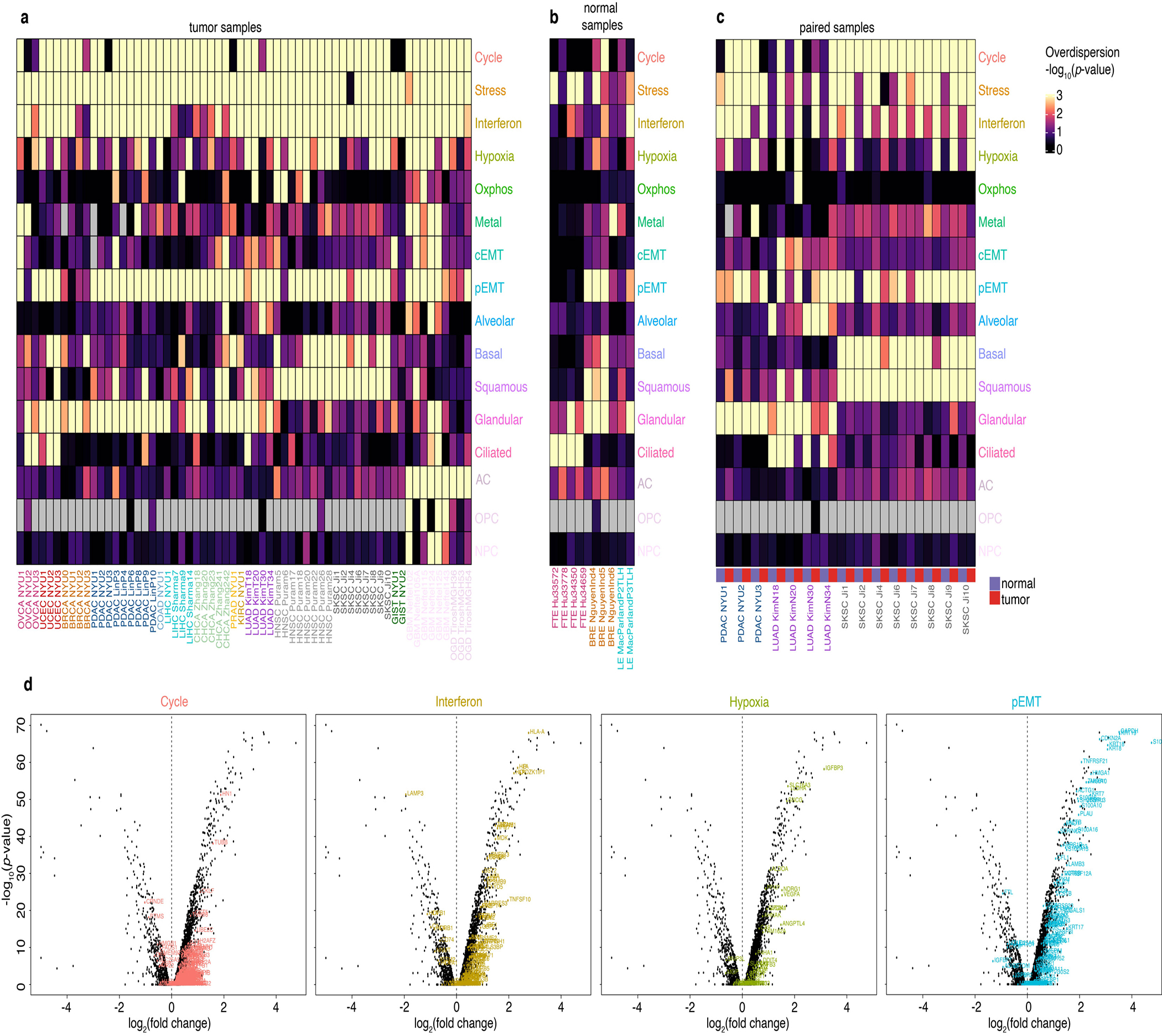
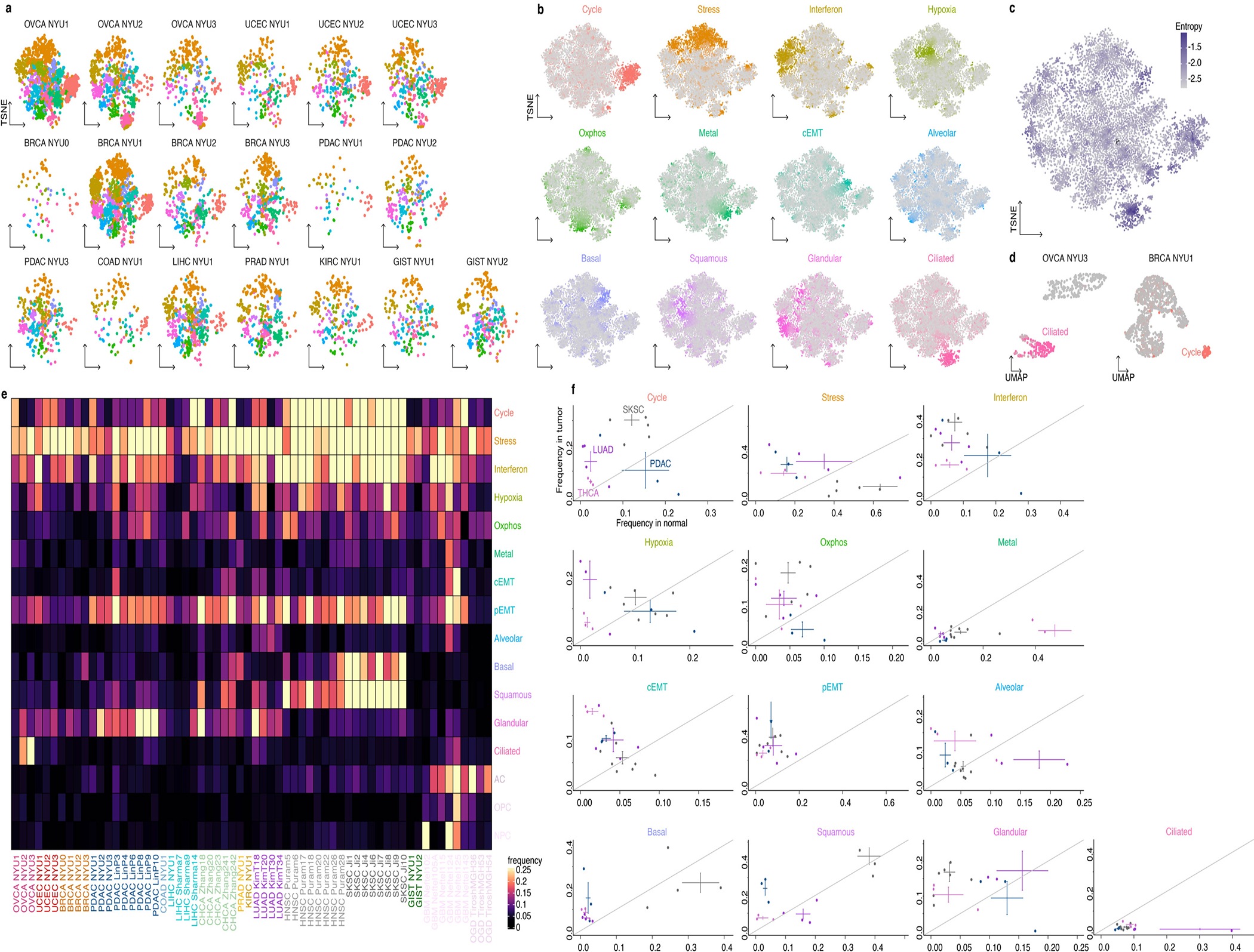
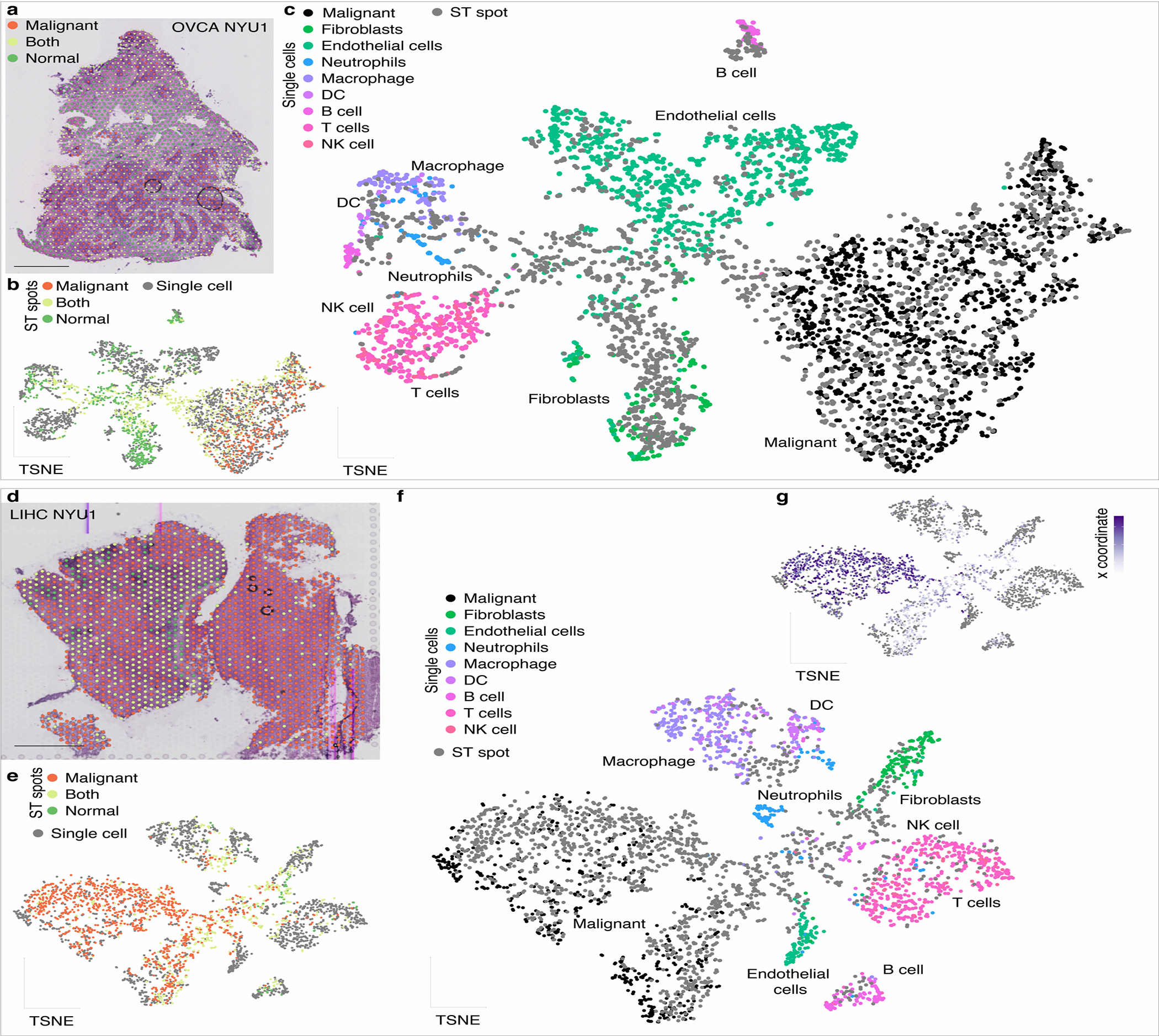
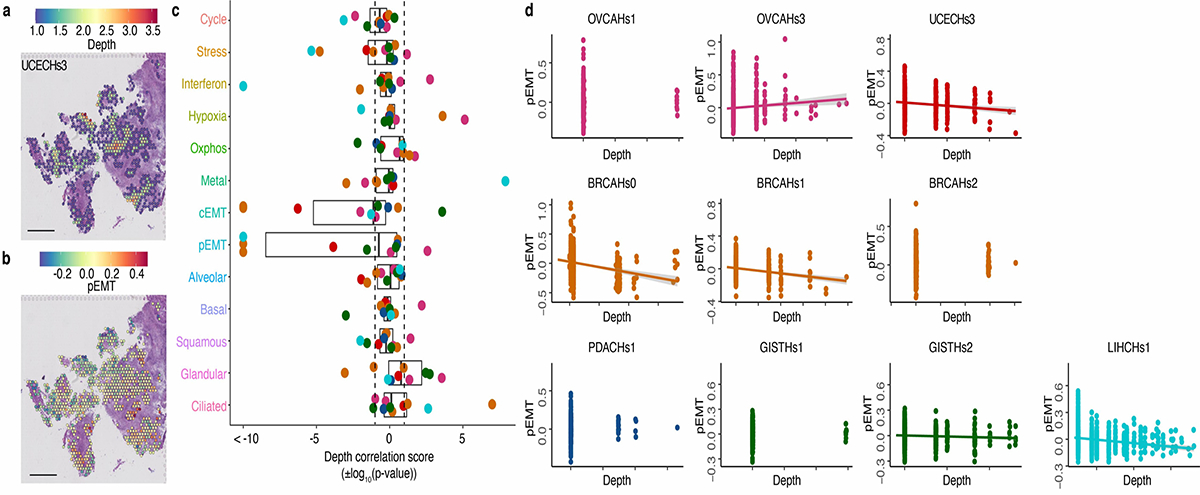
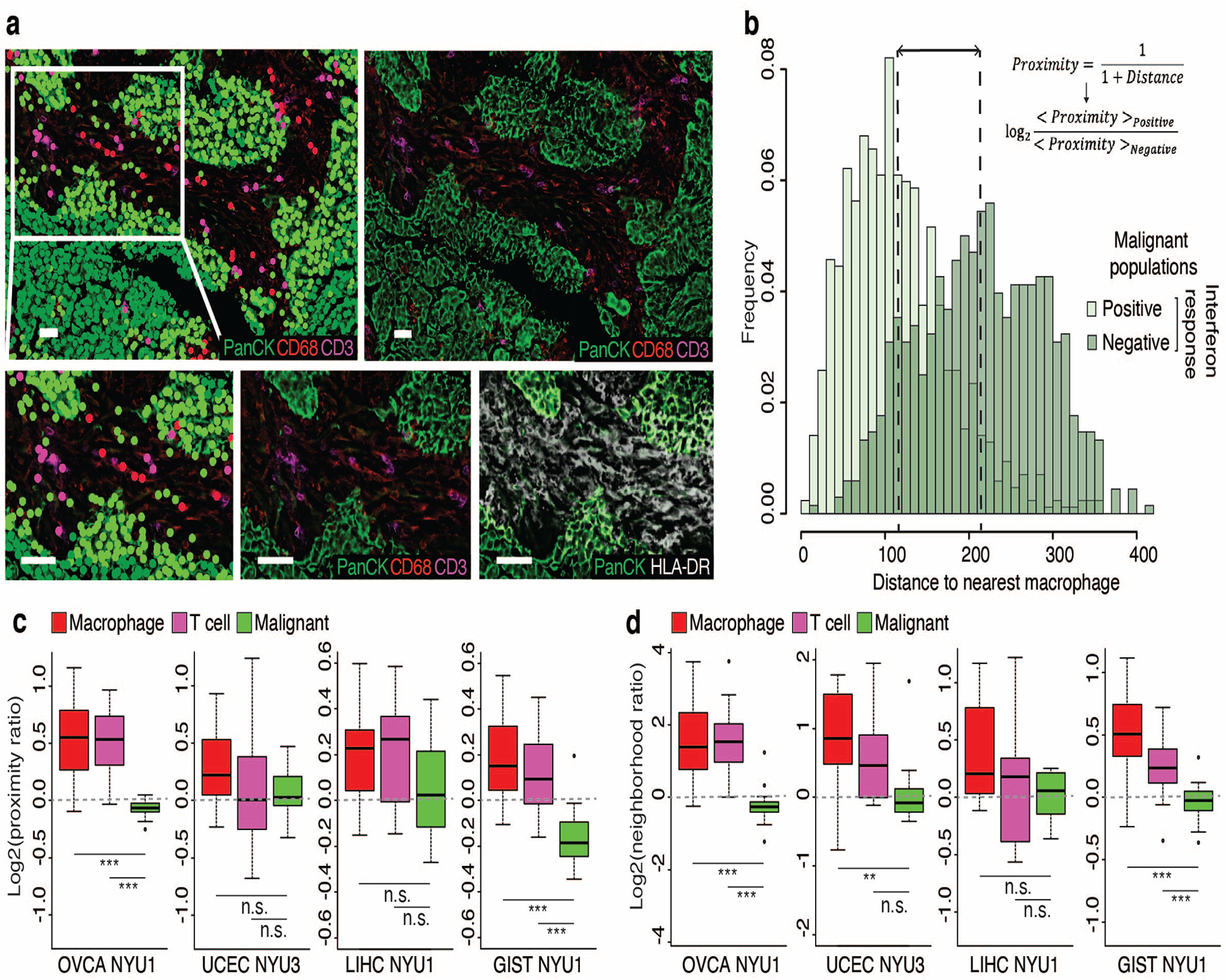
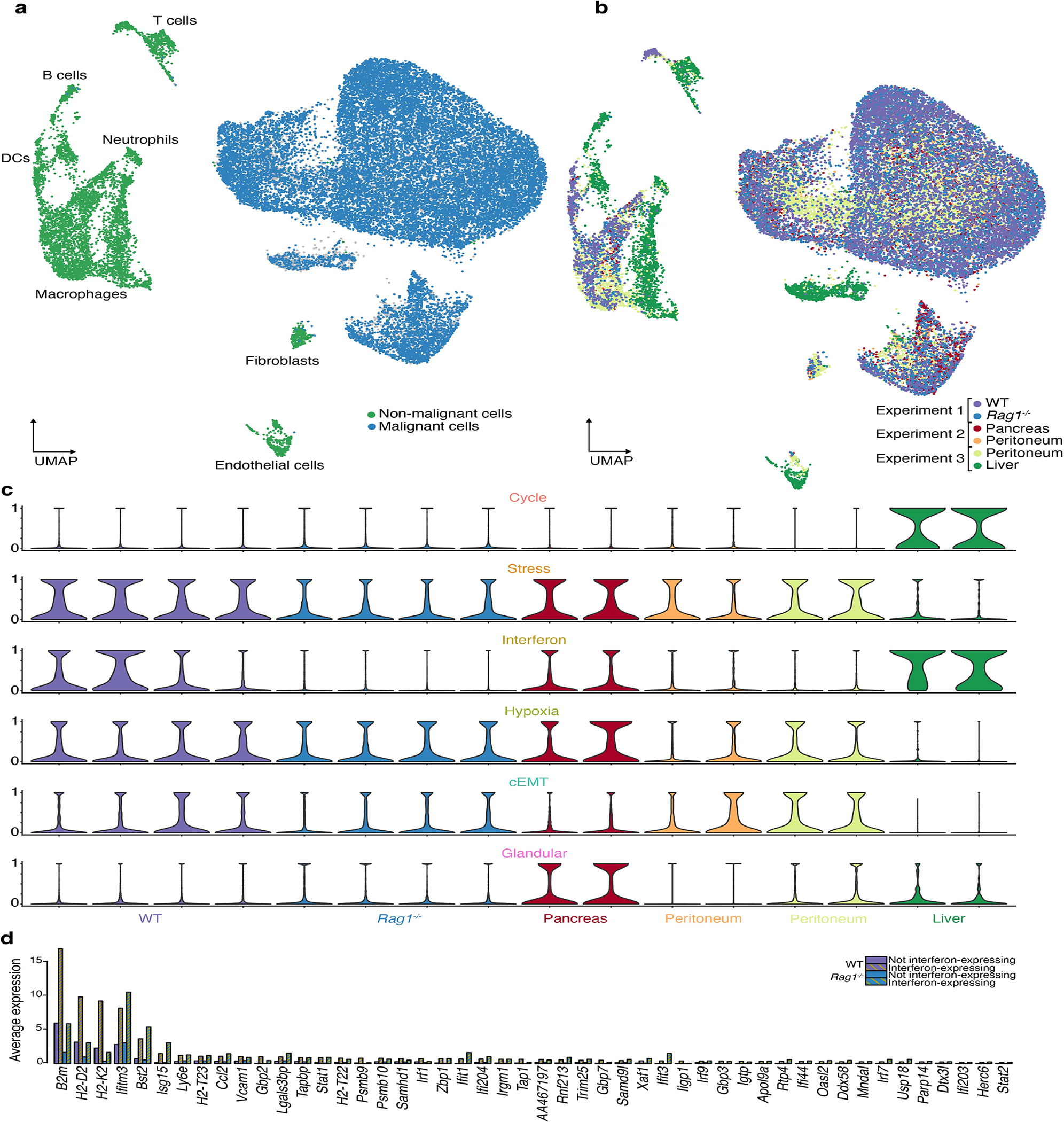
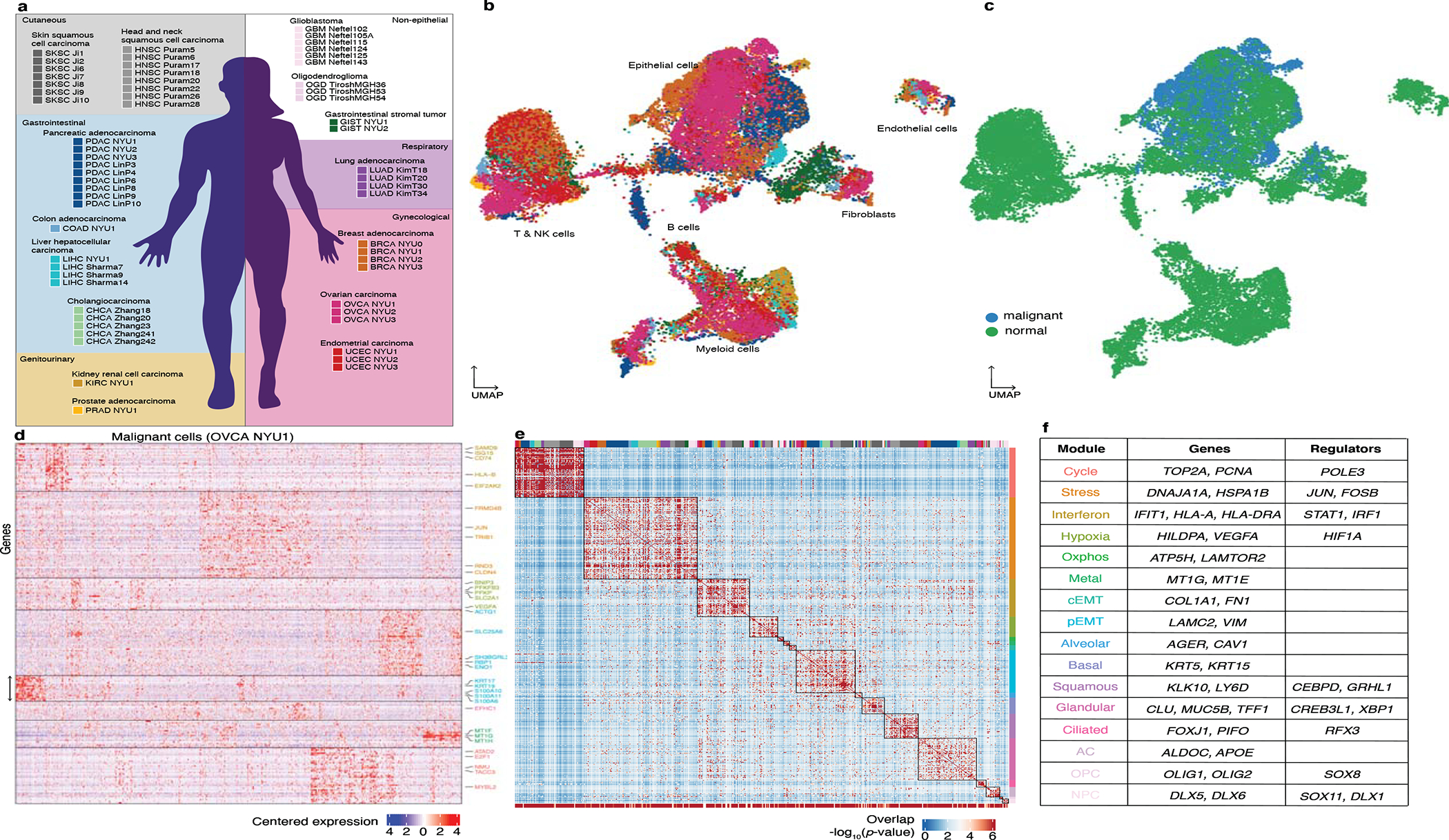
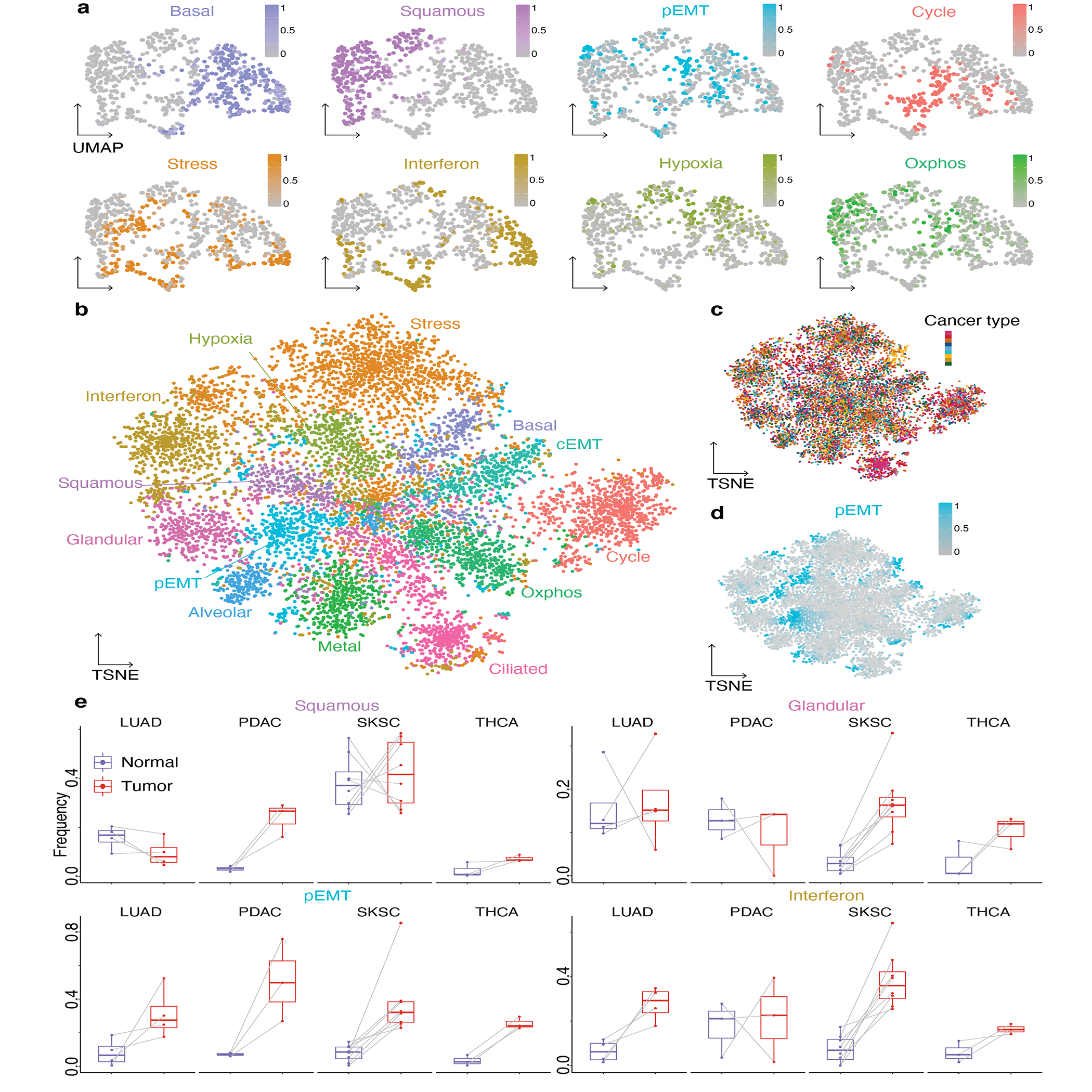
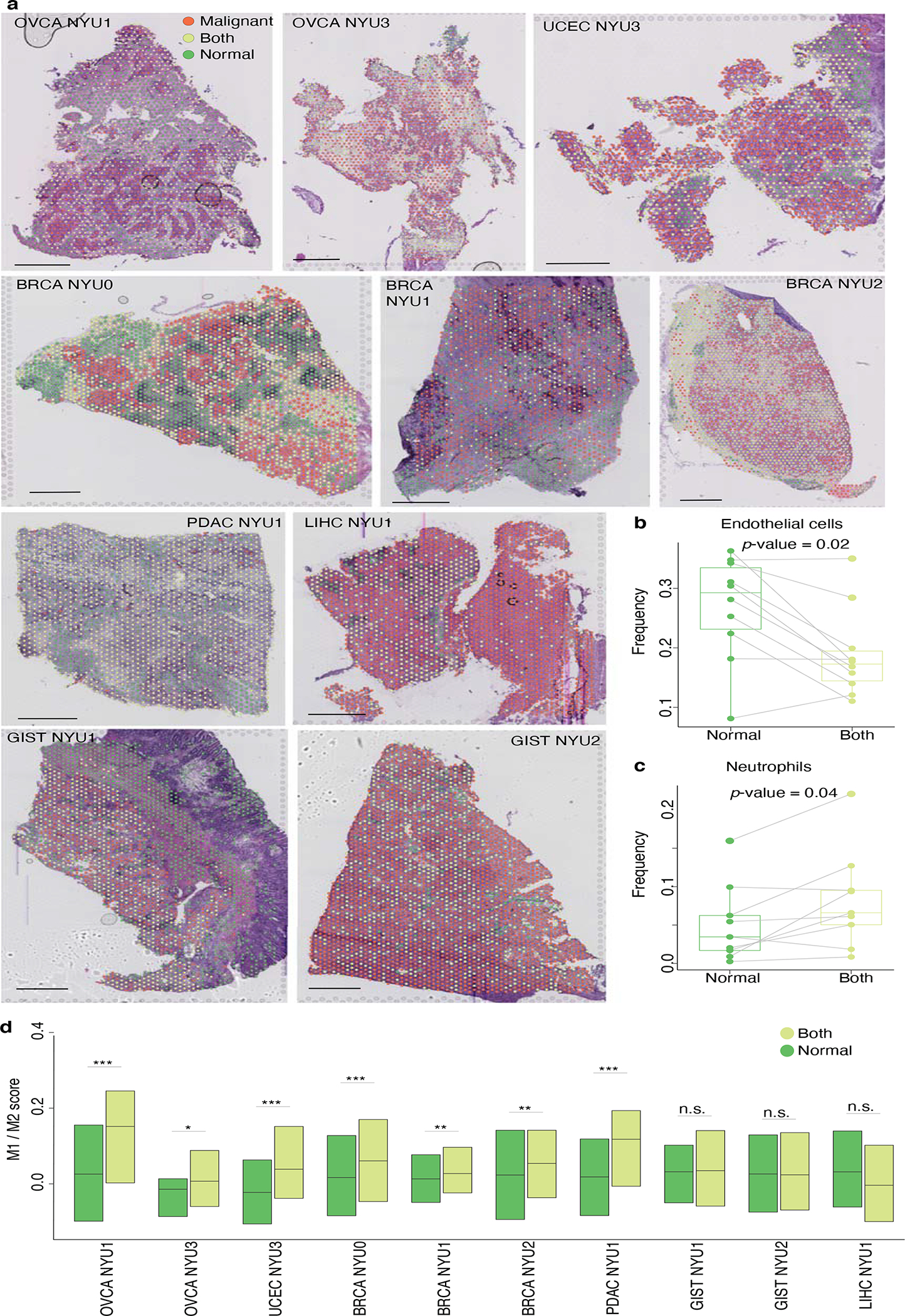
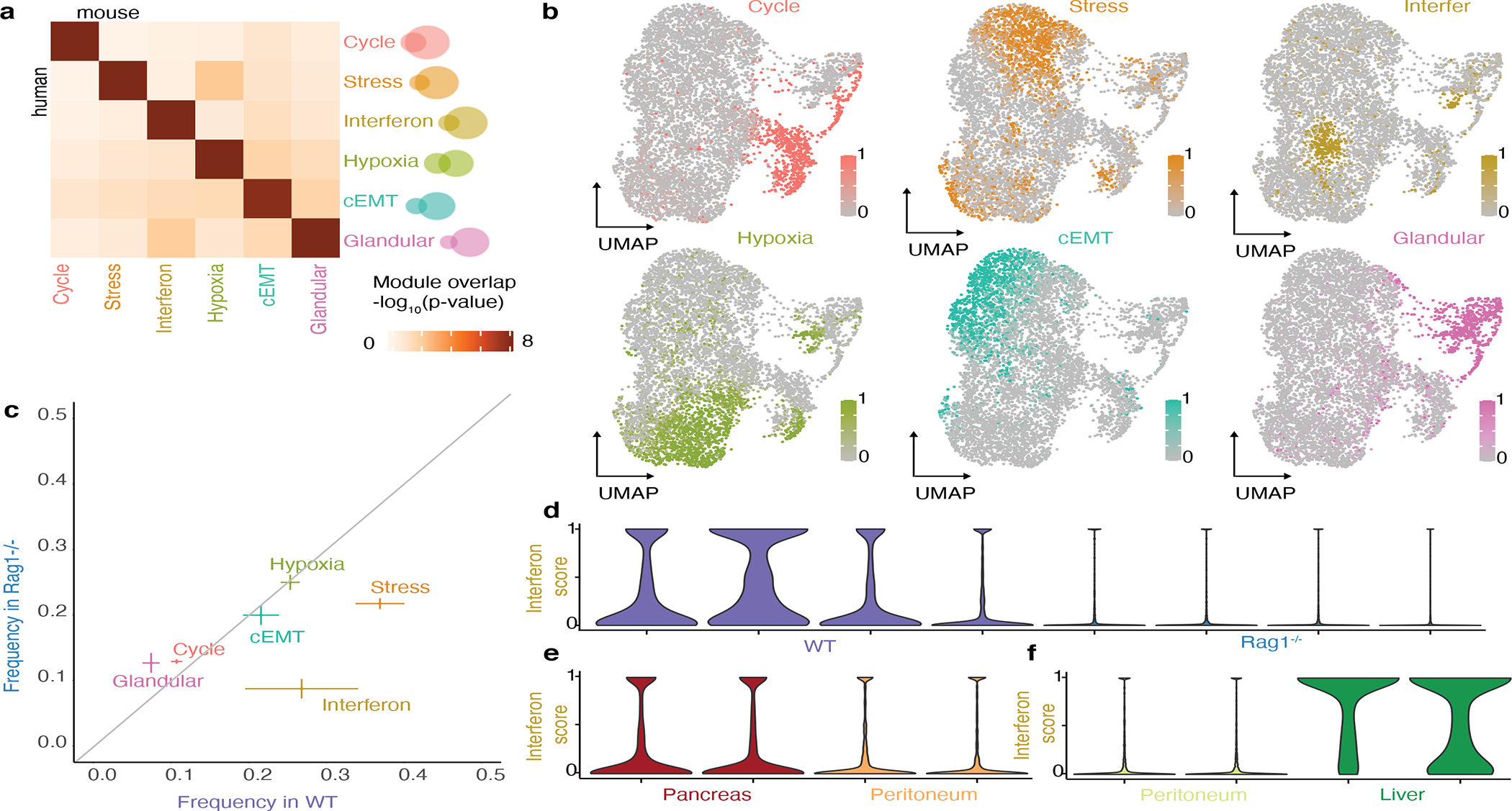
Transcriptional heterogeneity among malignant cells of a tumor has been studied in individual cancer types and shown to be organized into cancer cell states; however, it remains unclear to what extent these states span tumor types, constituting general features of cancer. Here, we perform a pan-cancer single-cell RNA-sequencing analysis across 15 cancer types and identify a catalog of gene modules whose expression defines recurrent cancer cell states including 'stress', 'interferon response', 'epithelial-mesenchymal transition', 'metal response', 'basal' and 'ciliated'. Spatial transcriptomic analysis linked the interferon response in cancer cells to T cells and macrophages in the tumor microenvironment. Using mouse models, we further found that induction of the interferon response module varies by tumor location and is diminished upon elimination of lymphocytes. Our work provides a framework for studying how cancer cell states interact with the tumor microenvironment to form organized systems capable of immune evasion, drug resistance and metastasis.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Figures



Comment in
-
Mapping the cancer cell states conserved across solid tumors.Nat Genet. 2022 Aug;54(8):1066-1067. doi: 10.1038/s41588-022-01151-7. Nat Genet. 2022. PMID: 35931862 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
