

Genetics of the human microglia regulome refines Alzheimer's disease risk loci
- PMID: 35931864
- PMCID: PMC9388367
- DOI: 10.1038/s41588-022-01149-1
Genetics of the human microglia regulome refines Alzheimer's disease risk loci
Abstract
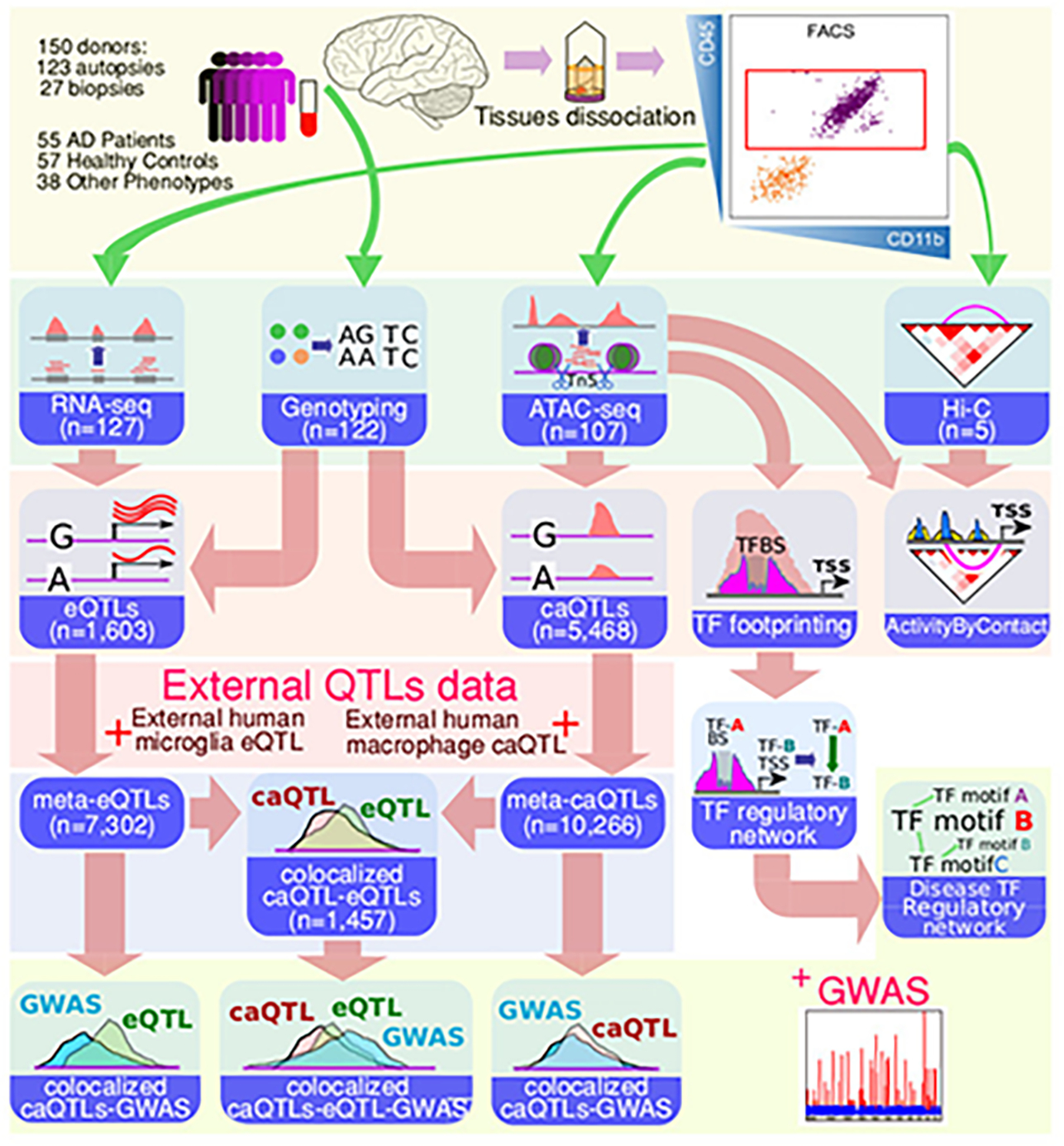
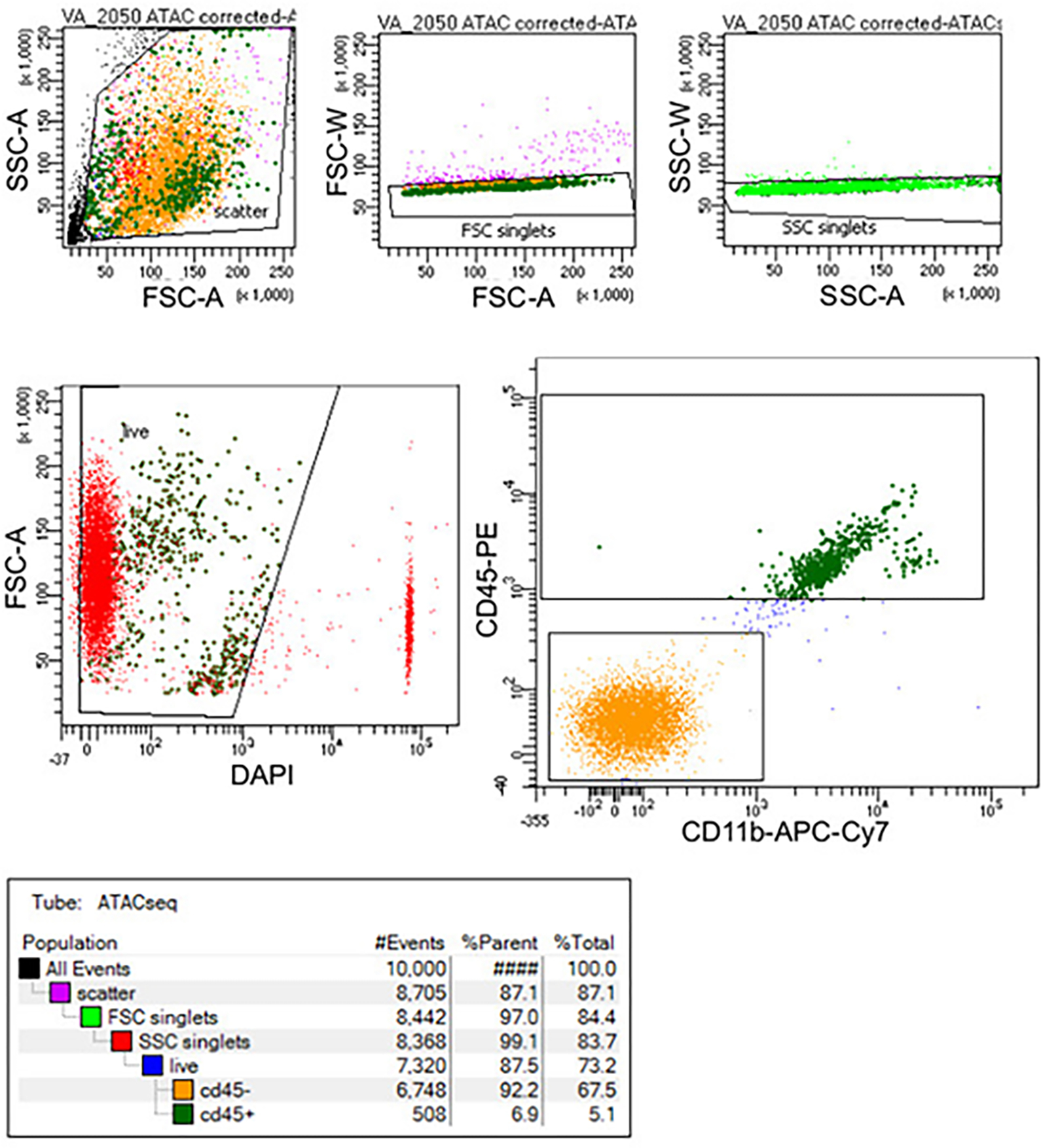
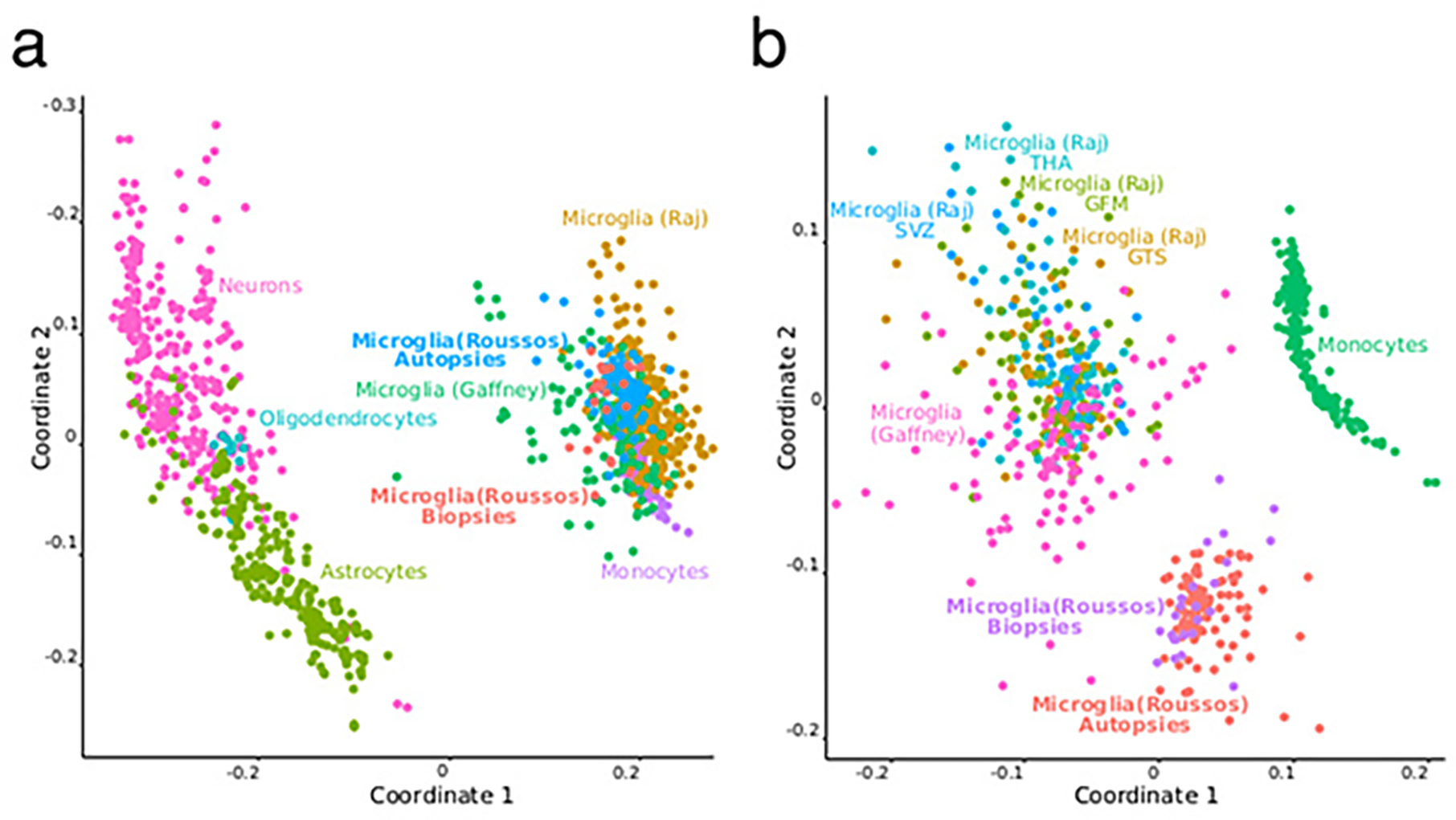
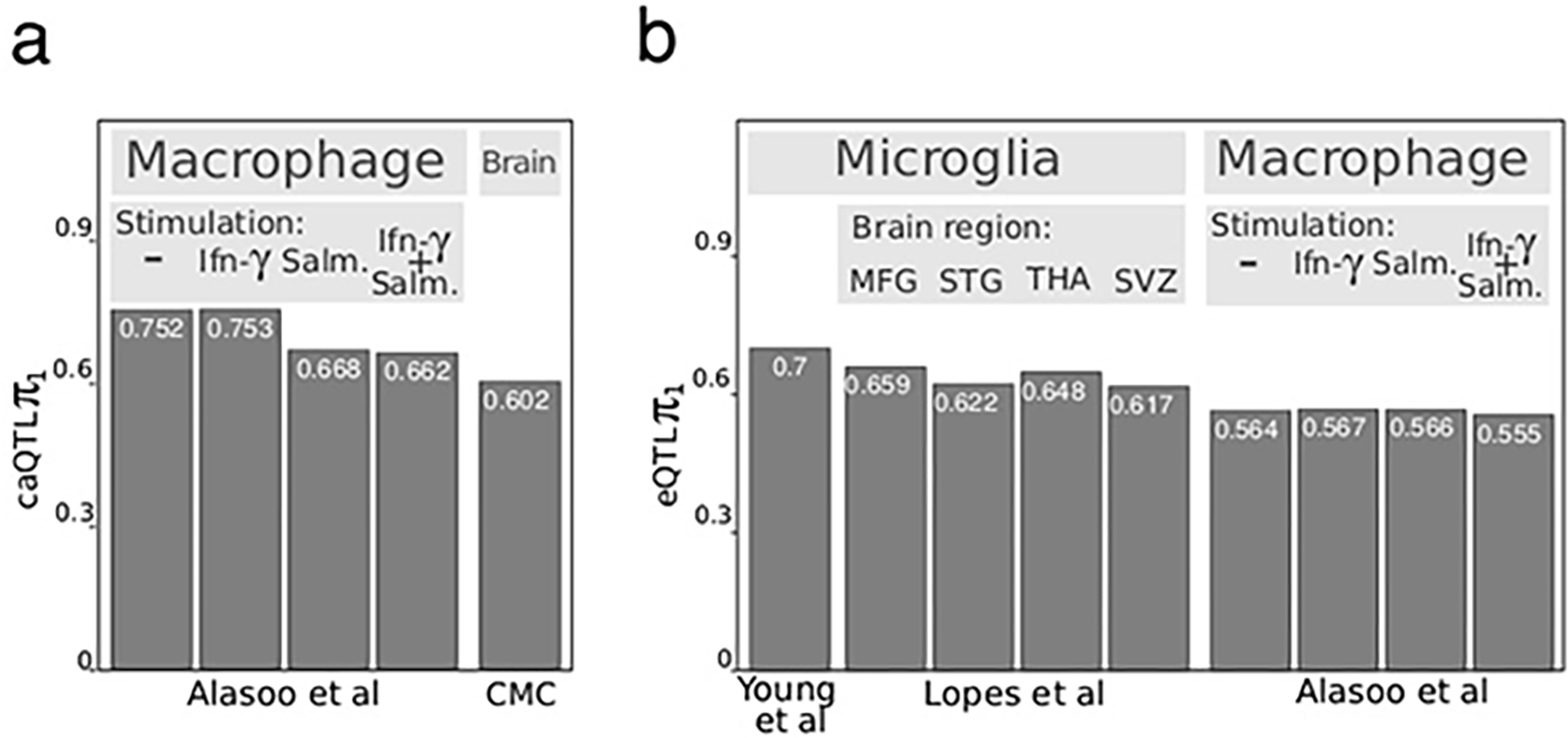
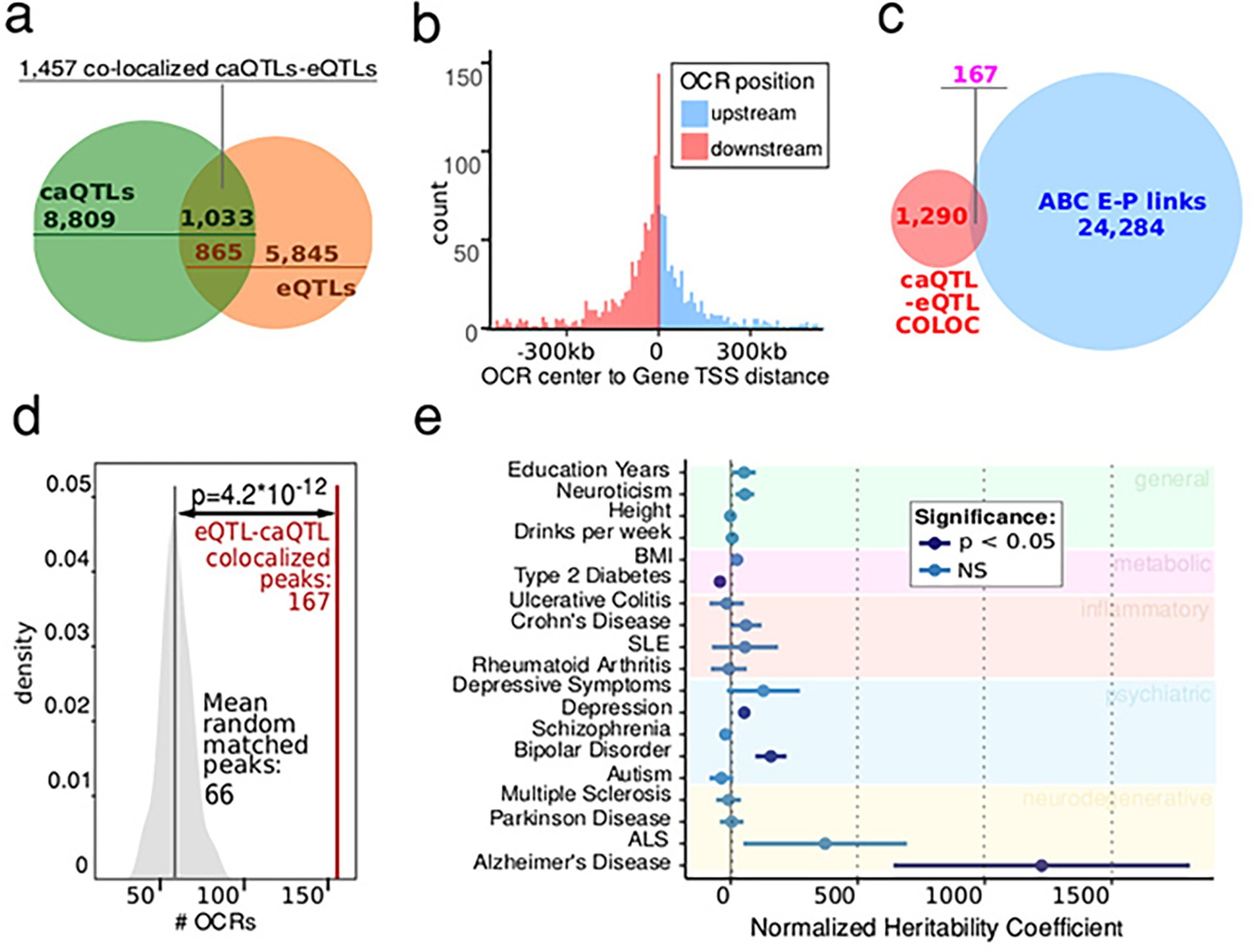
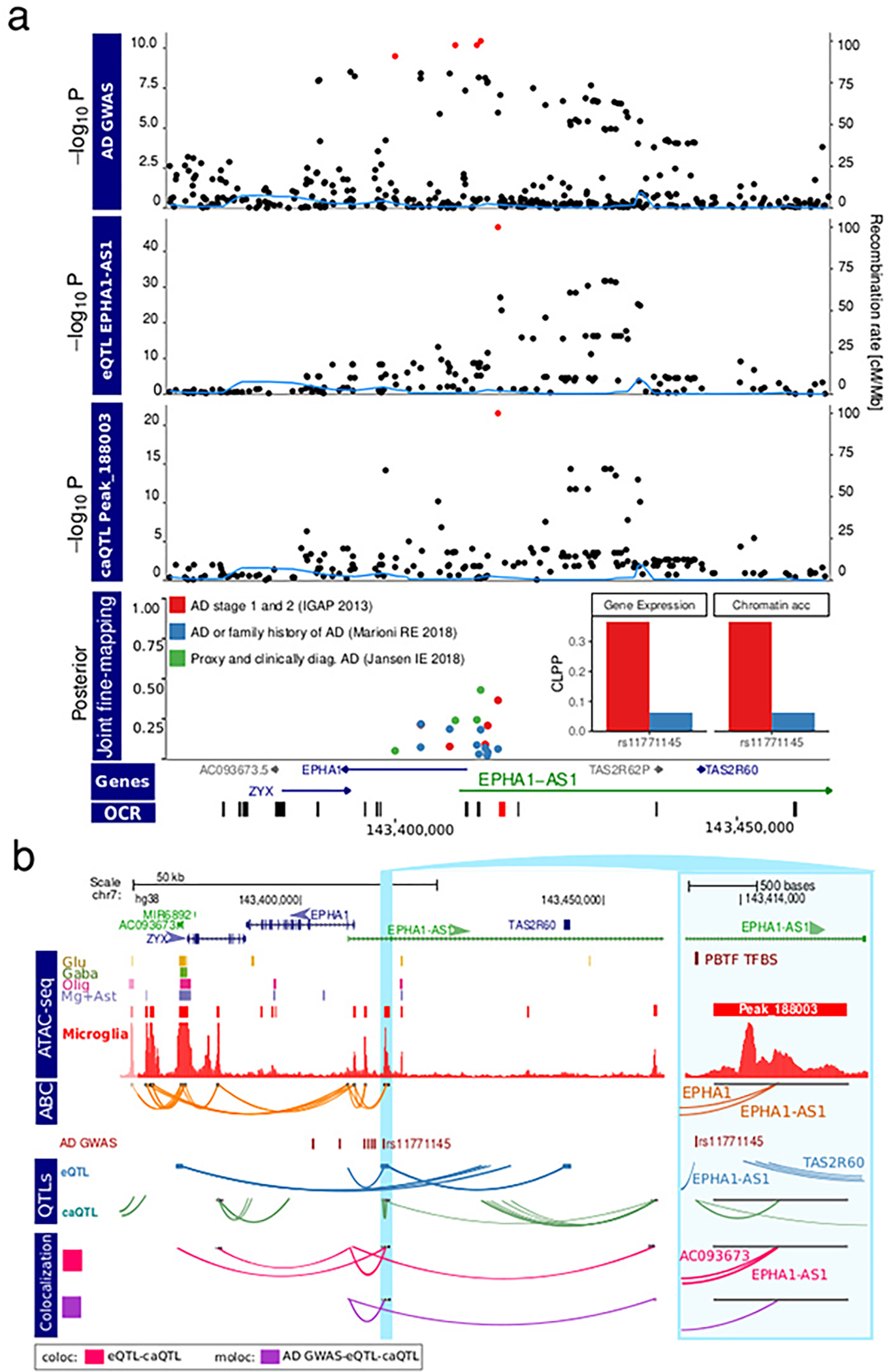
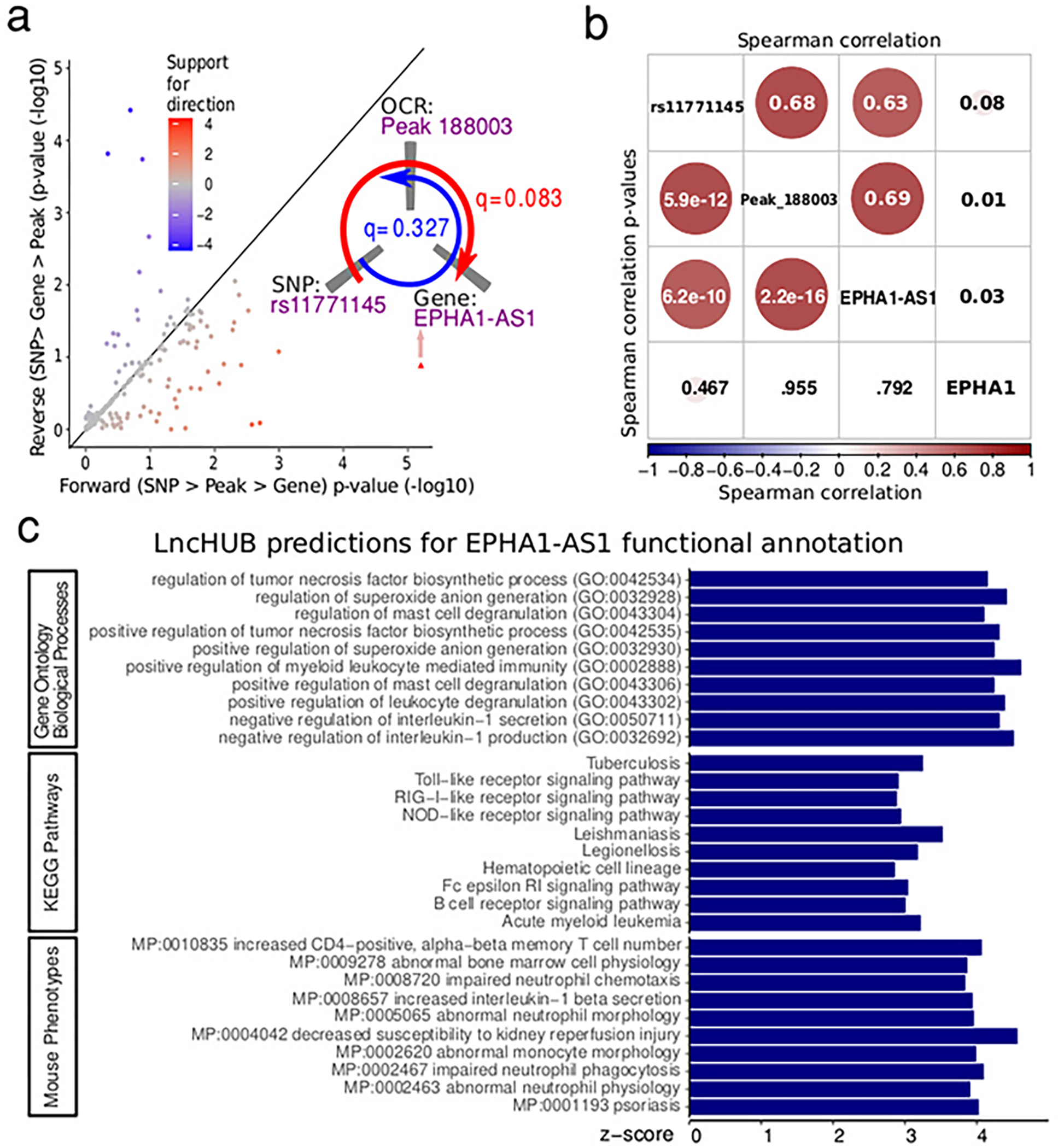
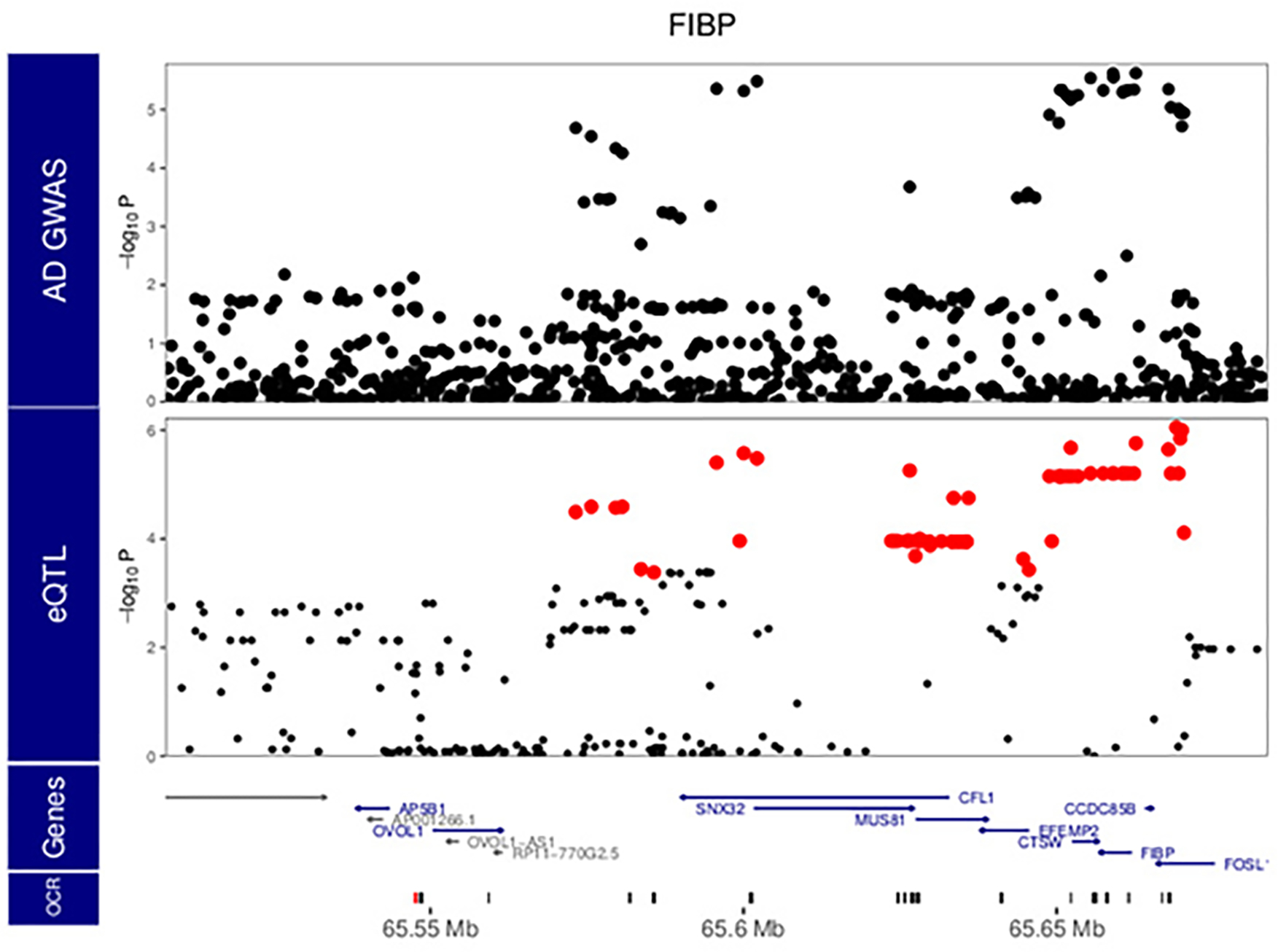
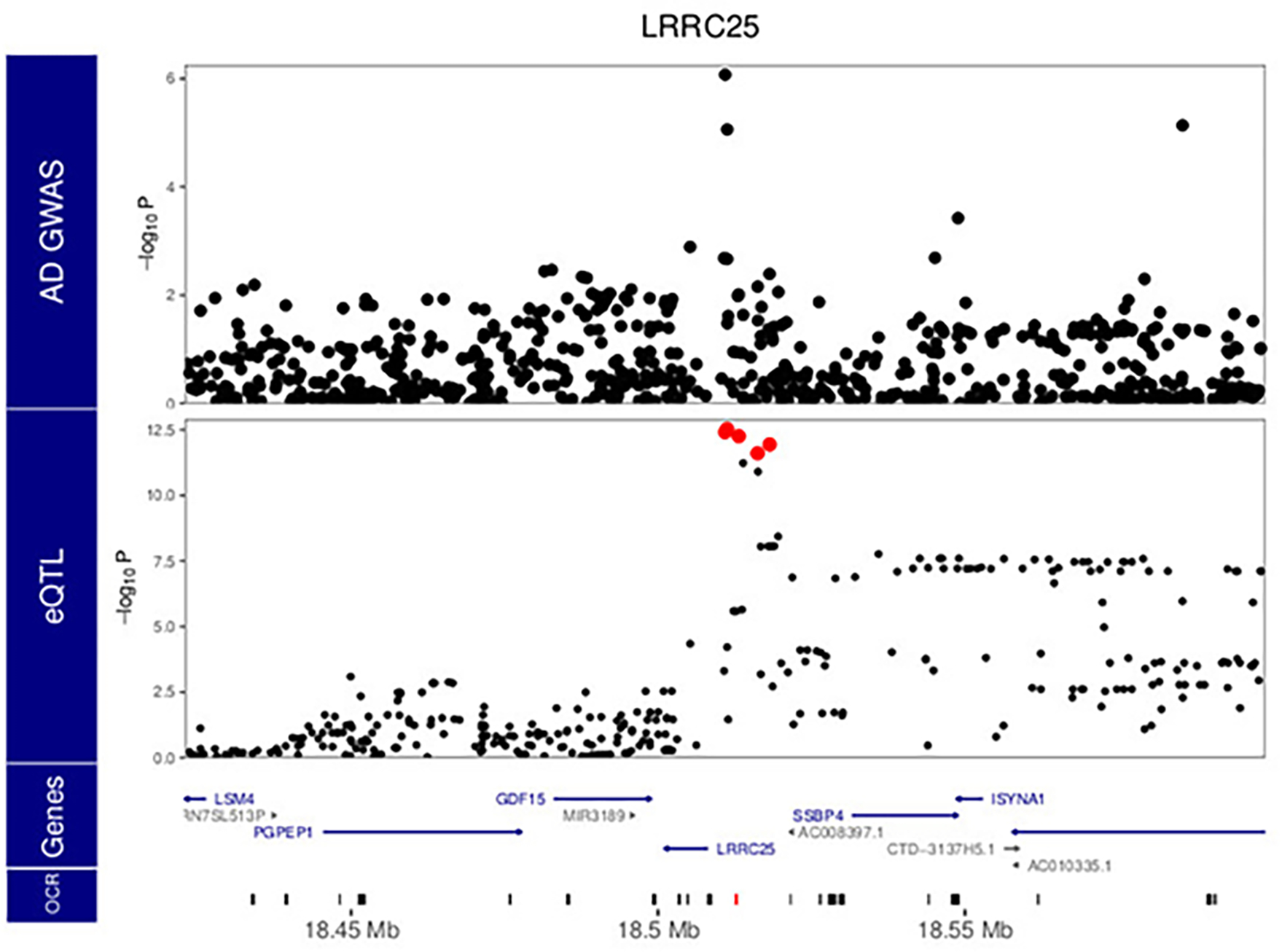
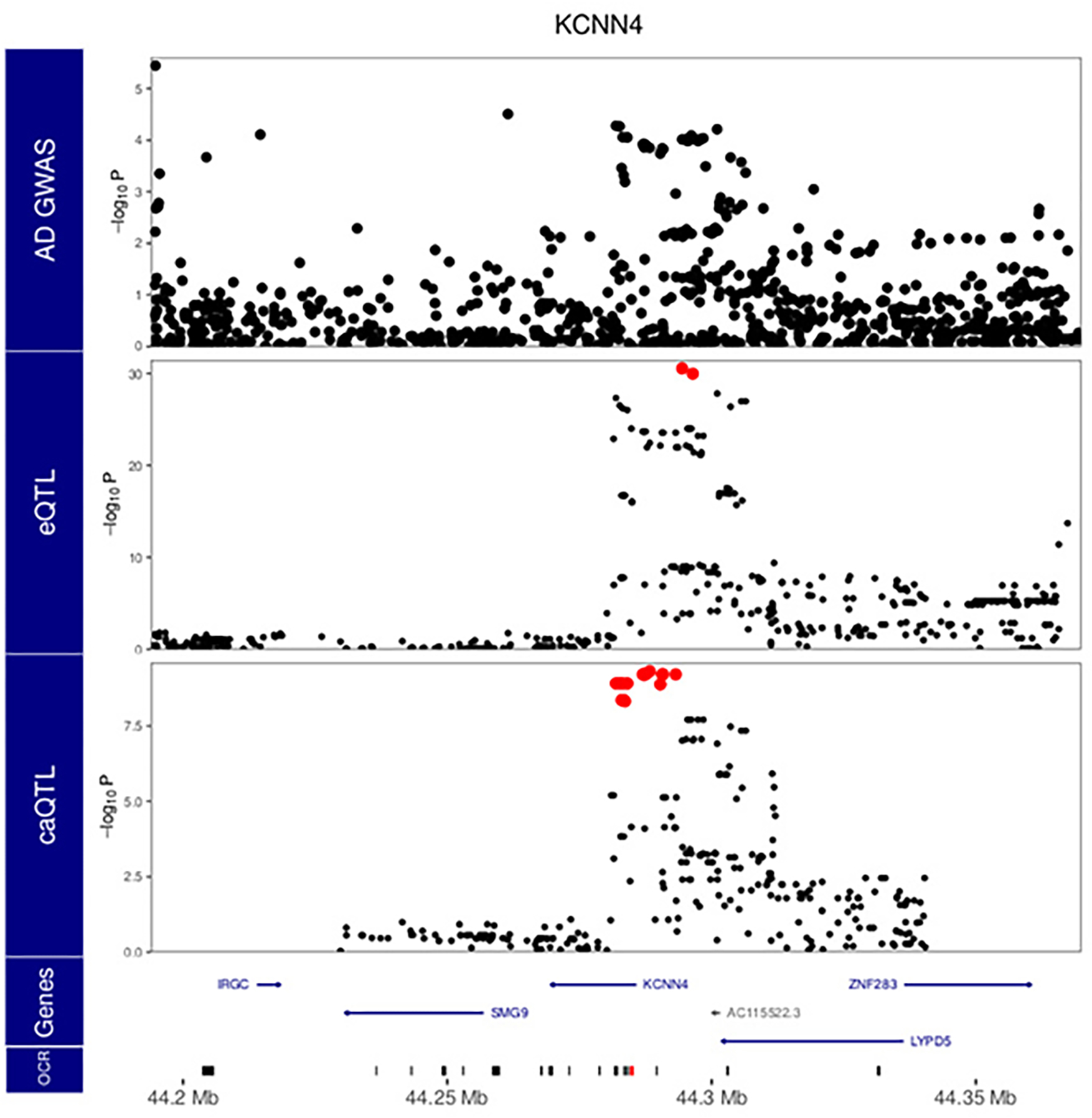
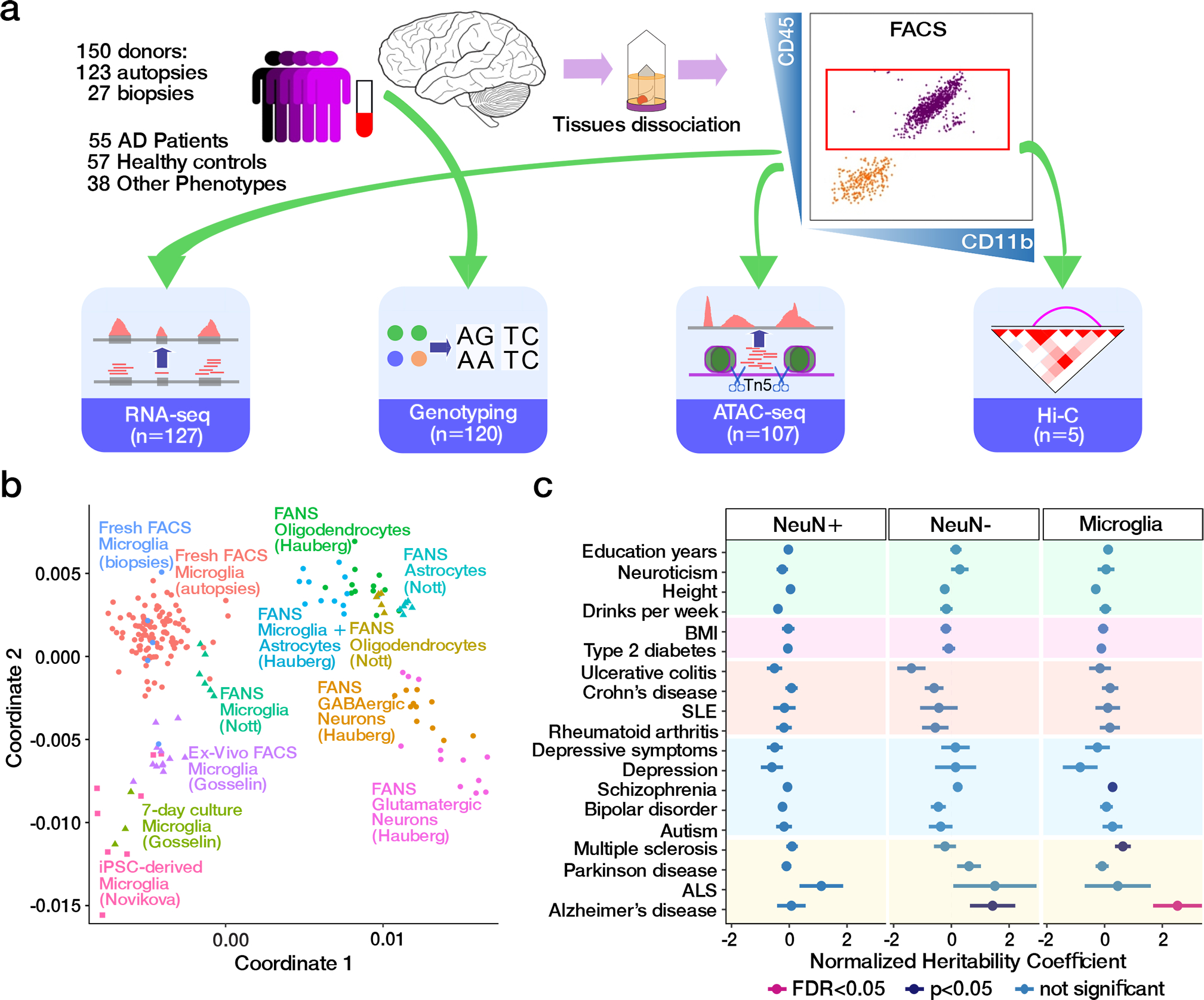
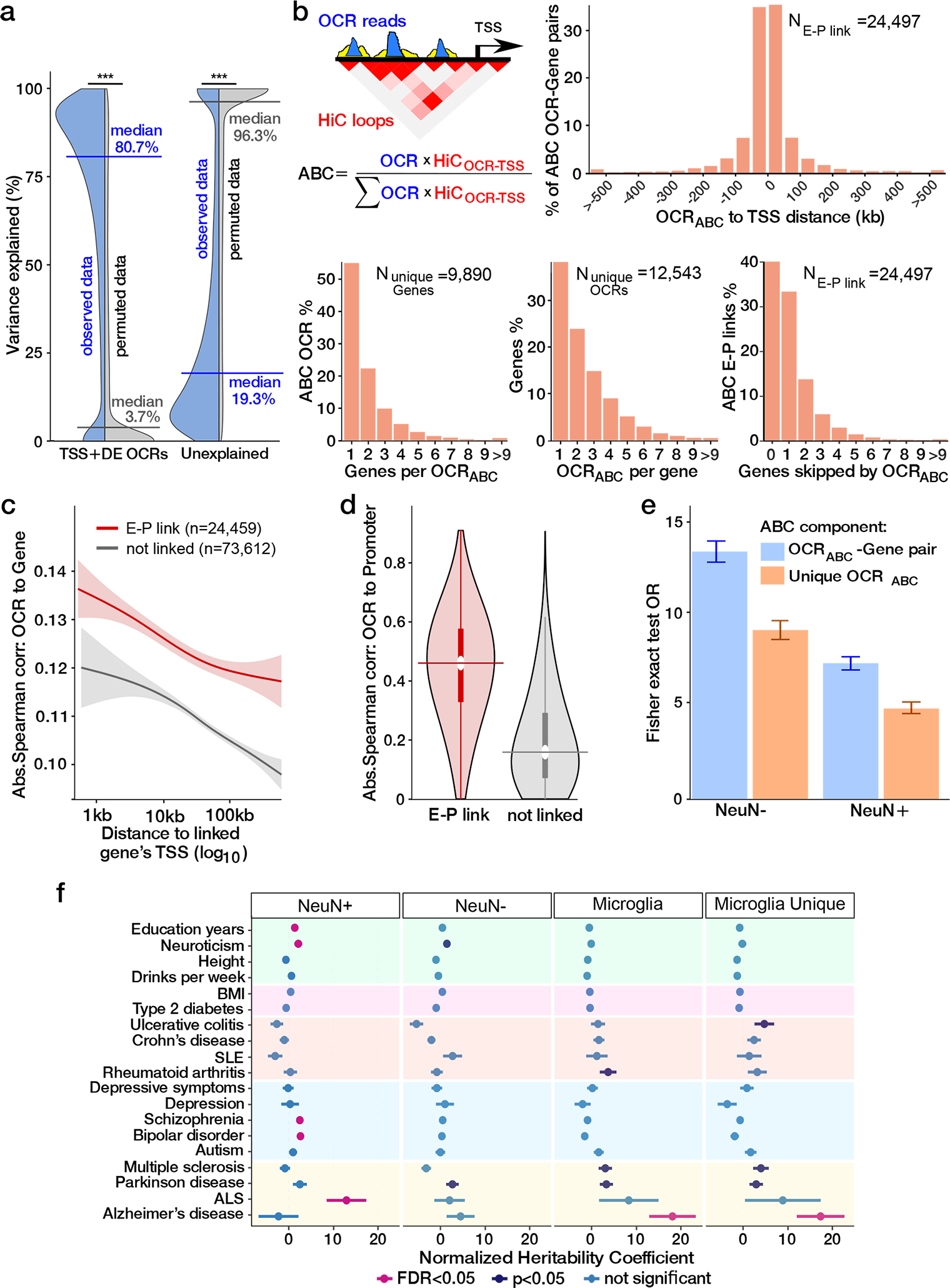
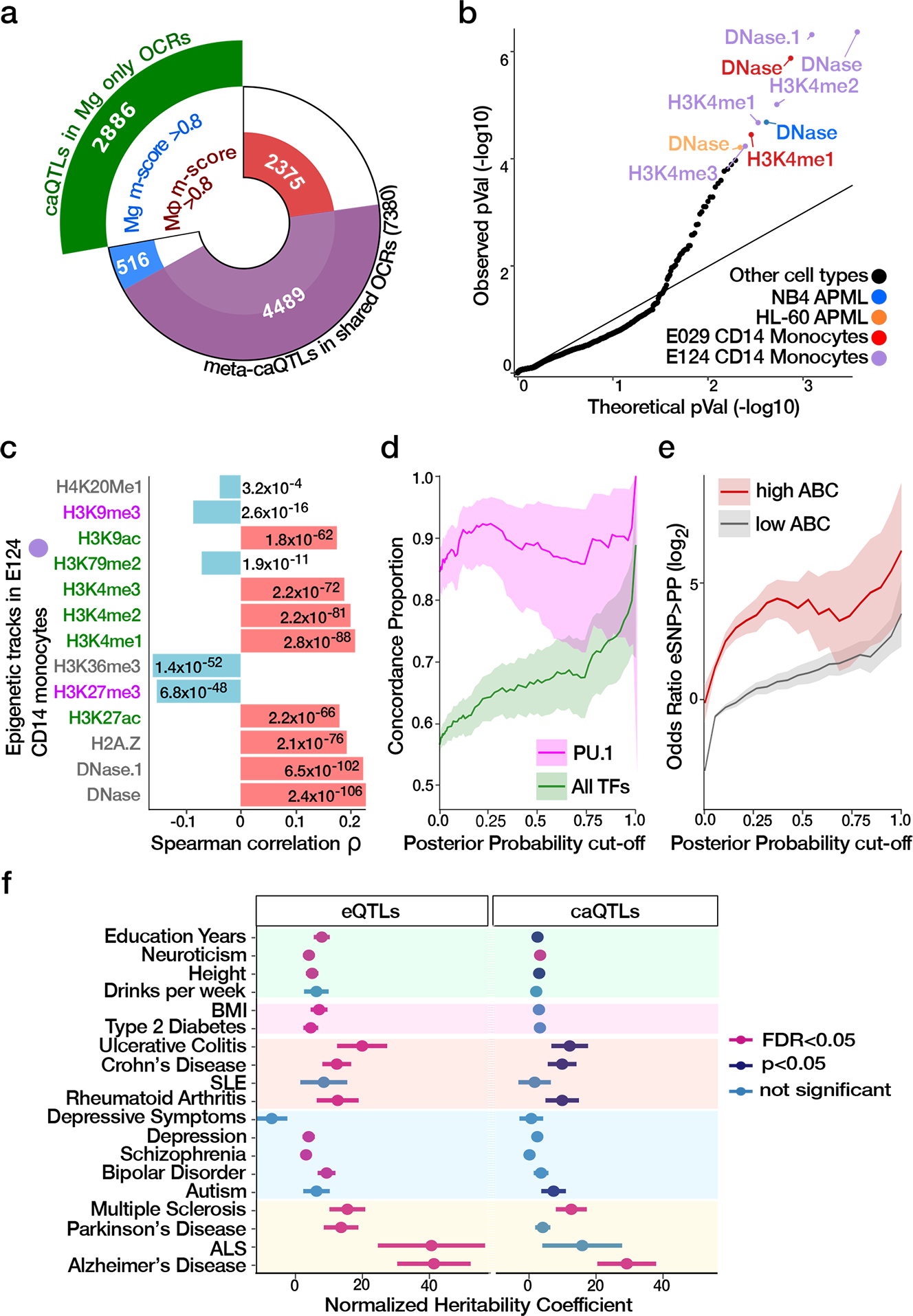
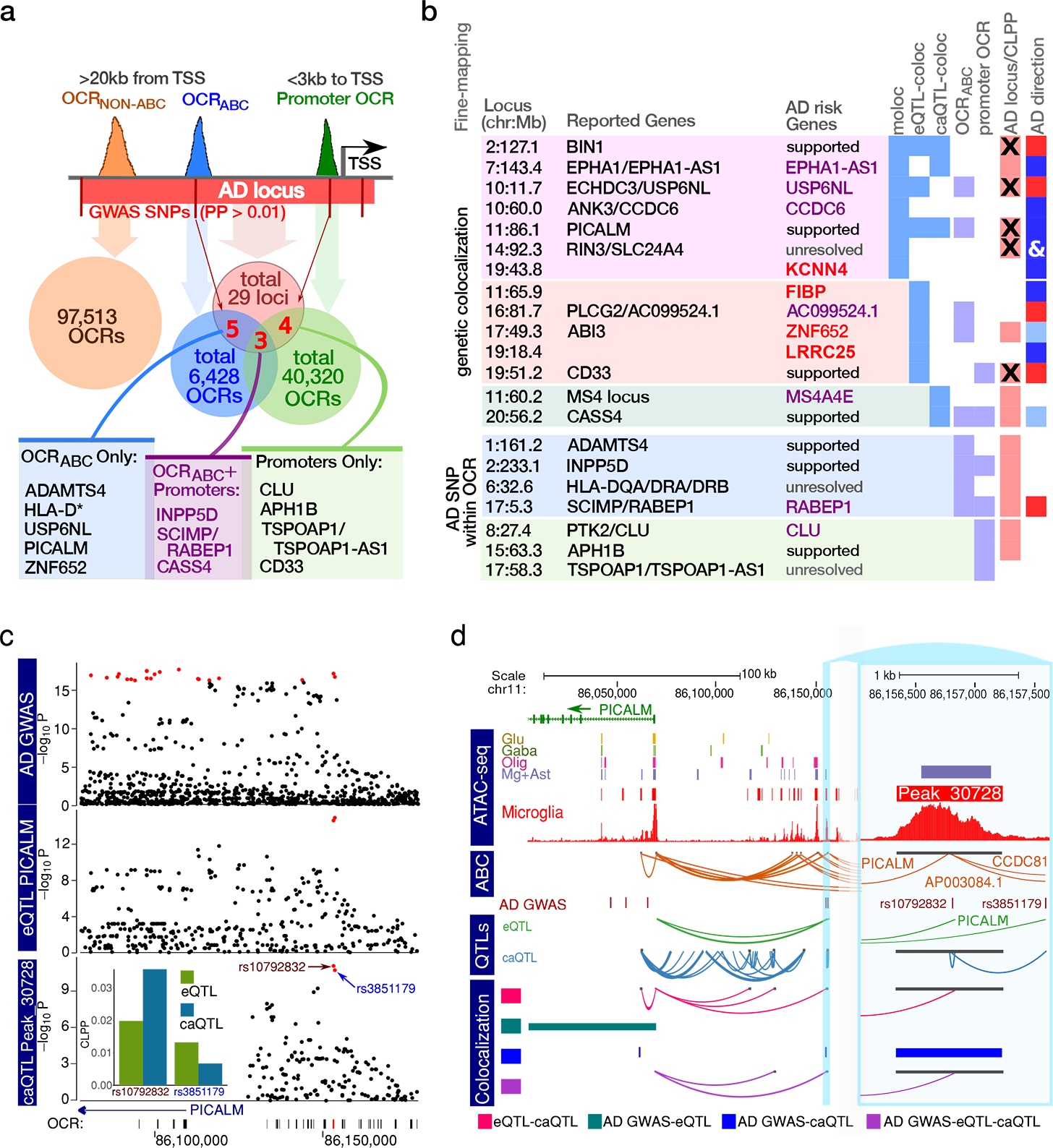
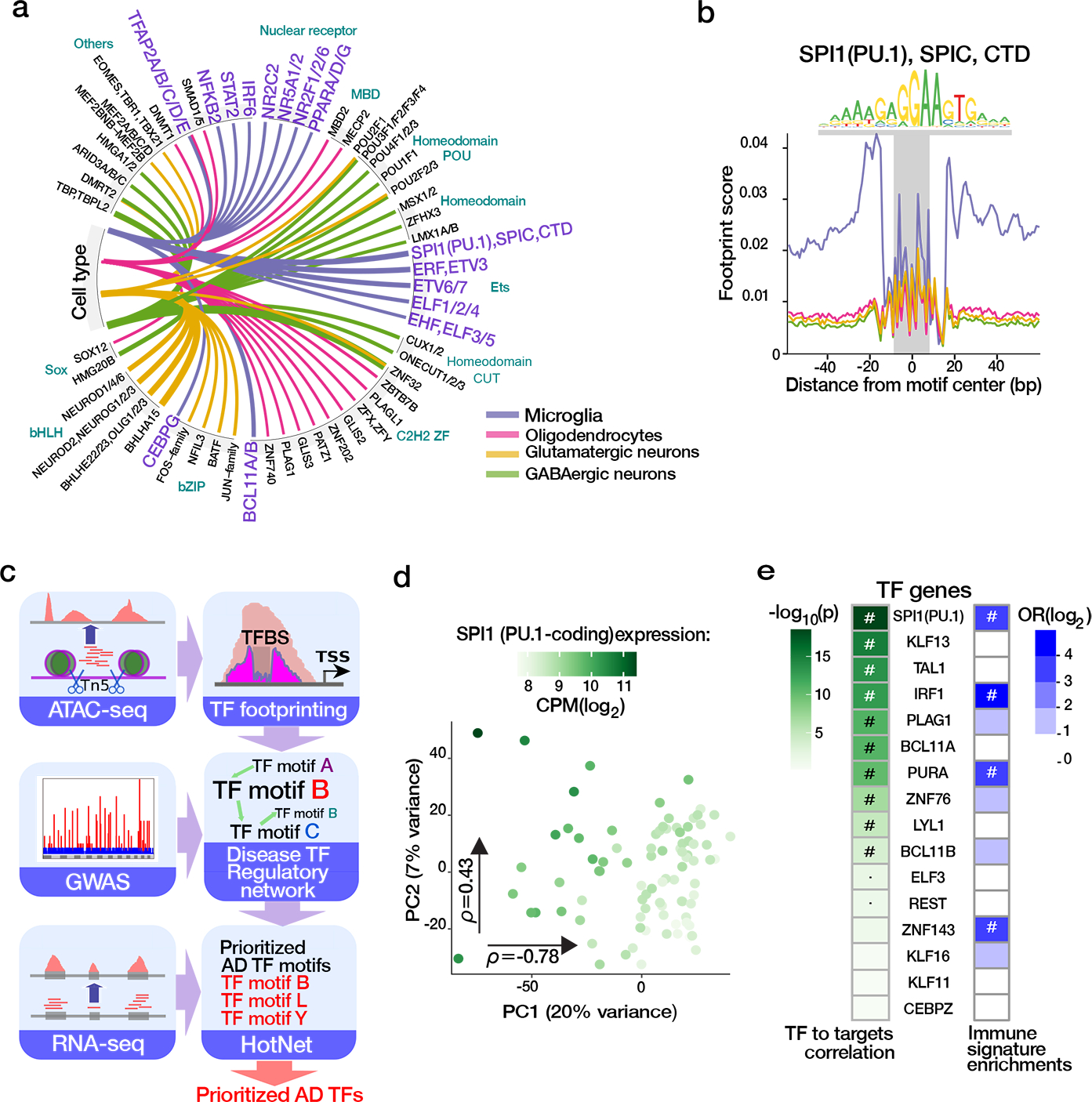
Microglia are brain myeloid cells that play a critical role in neuroimmunity and the etiology of Alzheimer's disease (AD), yet our understanding of how the genetic regulatory landscape controls microglial function and contributes to AD is limited. Here, we performed transcriptome and chromatin accessibility profiling in primary human microglia from 150 donors to identify genetically driven variation and cell-specific enhancer-promoter (E-P) interactions. Integrative fine-mapping analysis identified putative regulatory mechanisms for 21 AD risk loci, of which 18 were refined to a single gene, including 3 new candidate risk genes (KCNN4, FIBP and LRRC25). Transcription factor regulatory networks captured AD risk variation and identified SPI1 as a key putative regulator of microglia expression and AD risk. This comprehensive resource capturing variation in the human microglia regulome provides insights into the etiology of neurodegenerative disease.
© 2022. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Figures
References
-
- Salter MW & Stevens B Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027 (2017). - PubMed
-
- Bartels T, De Schepper S & Hong S Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 370, 66–69 (2020). - PubMed
-
- Song WM & Colonna M The identity and function of microglia in neurodegeneration. Nat. Immunol. 19, 1048–1058 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 AG072975/AG/NIA NIH HHS/United States
- RF1 AG036042/AG/NIA NIH HHS/United States
- R01 AG054005/AG/NIA NIH HHS/United States
- R01 AG069976/AG/NIA NIH HHS/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- S10 OD026880/OD/NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- R01 AG067025/AG/NIA NIH HHS/United States
- U01 AG068880/AG/NIA NIH HHS/United States
- S10 OD030463/OD/NIH HHS/United States
- R01 AG065582/AG/NIA NIH HHS/United States
- P30 AG066514/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
- R01 AG050986/AG/NIA NIH HHS/United States
- R01 AG065926/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
