

Novel copy number variation of COLQ gene in a Moroccan patient with congenital myasthenic syndrome: a case report and review of the literature
- PMID: 35932018
- PMCID: PMC9354381
- DOI: 10.1186/s12883-022-02822-y
Novel copy number variation of COLQ gene in a Moroccan patient with congenital myasthenic syndrome: a case report and review of the literature
Abstract
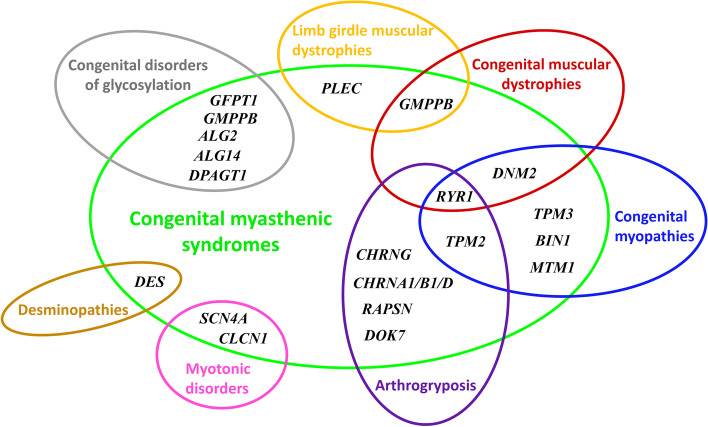
Background: Congenital myasthenic syndromes (CMSs) are rare genetic diseases due to abnormalities of the neuromuscular junction leading to permanent or transient muscle fatigability and weakness. To date, 32 genes were found to be involved in CMSs with autosomal dominant and/or recessive inheritance patterns. CMS with acetylcholinesterase deficiency, in particular, was determined to be due to biallelic mutations of COLQ gene with early-onset clinical signs. Here, we report clinical features and novel molecular findings of COLQ-related CMS in a Moroccan patient with a review of the literature for this rare form.
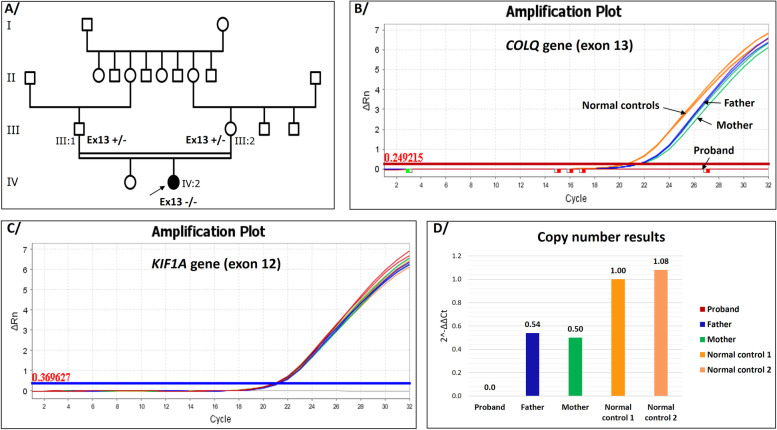
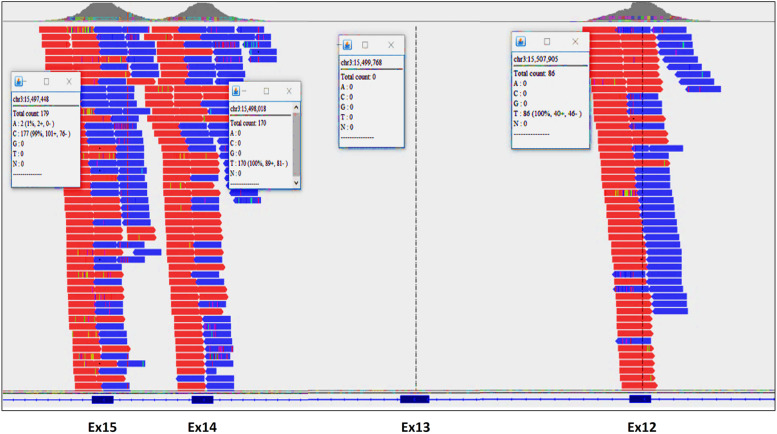
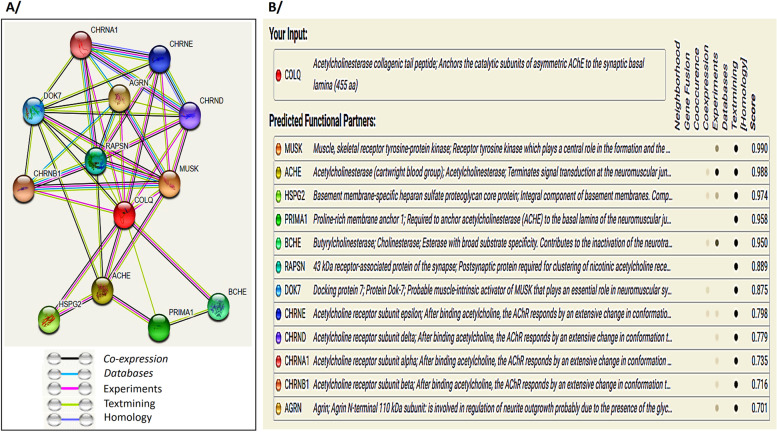
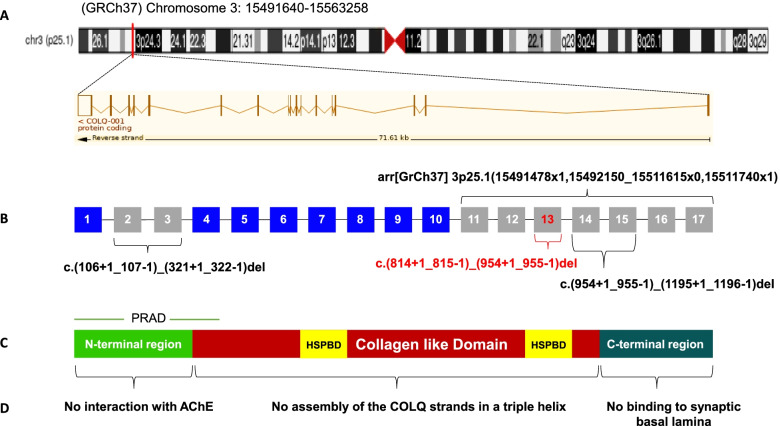
Case presentation: In this study, we report the case of a 28-month-old Moroccan female patient with hypotonia, associated to axial muscle weakness, global motor delay, bilateral ptosis, unilateral partial visual field deficiency with normal ocular motility, and fatigable muscle weakness. Clinical exome sequencing revealed a novel homozygous deletion of exon 13 in COLQ gene, NM_005677.4(COLQ):c.(814+1_815-1)_(954+1_955-1) del p.(Gly272Aspfs*11). This finding was subsequently confirmed by quantitative real-time PCR (qPCR) in the proband and her parents. In silico analysis of protein-protein interaction network by STRING tool revealed that 12 proteins are highly associated to COLQ with an elevated confidence score. Treatment with Salbutamol resulted in clear benefits and recovery.
Conclusions: This clinical observation illustrates the important place of next-generation sequencing in the precise molecular diagnosis of heterogeneous forms of CMS, the appropriate management and targeted treatment, and genetic counseling of families, with a better characterization of the mutational profile of this rare disease in the Moroccan population.
Keywords: COLQ gene; Case report; Congenital myasthenic syndrome; Novel CNV; Protein-protein interaction.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
The First Case of Congenital Myasthenic Syndrome Caused by a Large Homozygous Deletion in the C-Terminal Region of COLQ (Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase) Protein.Genes (Basel). 2020 Dec 18;11(12):1519. doi: 10.3390/genes11121519. Genes (Basel). 2020. PMID: 33353066 Free PMC article.
-
Clinical and molecular analysis of a novel COLQ missense mutation causing congenital myasthenic syndrome in a Syrian family.Pediatr Neurol. 2014 Jul;51(1):165-9. doi: 10.1016/j.pediatrneurol.2014.03.012. Epub 2014 Mar 22. Pediatr Neurol. 2014. PMID: 24938146
-
COLQ-Congenital myasthenic syndrome in an Iranian cohort: the clinical and genetics spectrum.Orphanet J Rare Dis. 2024 Mar 12;19(1):113. doi: 10.1186/s13023-024-03116-x. Orphanet J Rare Dis. 2024. PMID: 38475910 Free PMC article.
-
Pharmacological Treatments for Congenital Myasthenic Syndromes Caused by COLQ Mutations.Curr Neuropharmacol. 2023;21(7):1594-1605. doi: 10.2174/1570159X21666230126145652. Curr Neuropharmacol. 2023. PMID: 36703579 Free PMC article. Review.
-
Congenital myasthenic syndromes with acetylcholinesterase deficiency, the pathophysiological mechanisms.Ann N Y Acad Sci. 2018 Feb;1413(1):104-110. doi: 10.1111/nyas.13595. Ann N Y Acad Sci. 2018. PMID: 29405353 Review.
Cited by
-
Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes-A Comprehensive Review.Int J Mol Sci. 2023 Feb 13;24(4):3730. doi: 10.3390/ijms24043730. Int J Mol Sci. 2023. PMID: 36835142 Free PMC article. Review.
-
Identification and validation of key genes associated with atrial fibrillation in the elderly.Front Cardiovasc Med. 2023 Mar 29;10:1118686. doi: 10.3389/fcvm.2023.1118686. eCollection 2023. Front Cardiovasc Med. 2023. PMID: 37063972 Free PMC article.
-
COLQ-related congenital myasthenic syndrome: An integrative view.Neurogenetics. 2023 Jul;24(3):189-200. doi: 10.1007/s10048-023-00719-7. Epub 2023 May 25. Neurogenetics. 2023. PMID: 37231228
-
Expression assay of the COLQ in a family with congenital myasthenic syndrome and symptomatic carriers.Clin Case Rep. 2023 Oct 23;11(10):e8062. doi: 10.1002/ccr3.8062. eCollection 2023 Oct. Clin Case Rep. 2023. PMID: 37881193 Free PMC article.
-
A Pediatric Case of COLQ-Related Congenital Myasthenic Syndrome with Marked Fatigue.Children (Basel). 2023 Apr 24;10(5):769. doi: 10.3390/children10050769. Children (Basel). 2023. PMID: 37238317 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
