

Microglia-specific deletion of histone deacetylase 3 promotes inflammation resolution, white matter integrity, and functional recovery in a mouse model of traumatic brain injury
- PMID: 35933343
- PMCID: PMC9357327
- DOI: 10.1186/s12974-022-02563-2
Microglia-specific deletion of histone deacetylase 3 promotes inflammation resolution, white matter integrity, and functional recovery in a mouse model of traumatic brain injury
Abstract
Background: Histone deacetylases (HDACs) are believed to exacerbate traumatic brain injury (TBI) based on studies using pan-HDAC inhibitors. However, the HDAC isoform responsible for the detrimental effects and the cell types involved remain unknown, which may hinder the development of specific targeting strategies that boost therapeutic efficacy while minimizing side effects. Microglia are important mediators of post-TBI neuroinflammation and critically impact TBI outcome. HDAC3 was reported to be essential to the inflammatory program of in vitro cultured macrophages, but its role in microglia and in the post-TBI brain has not been investigated in vivo.
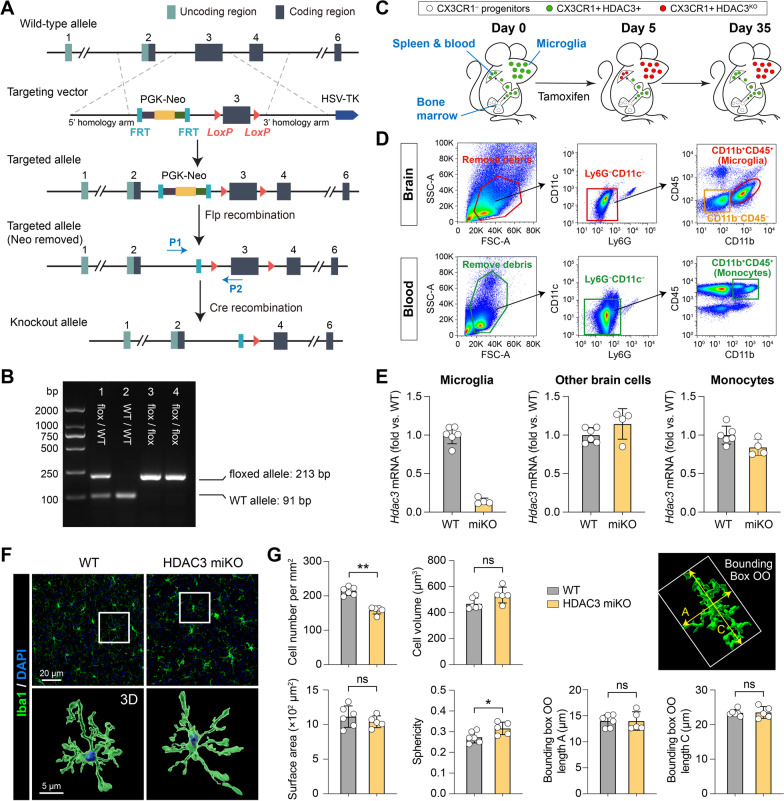
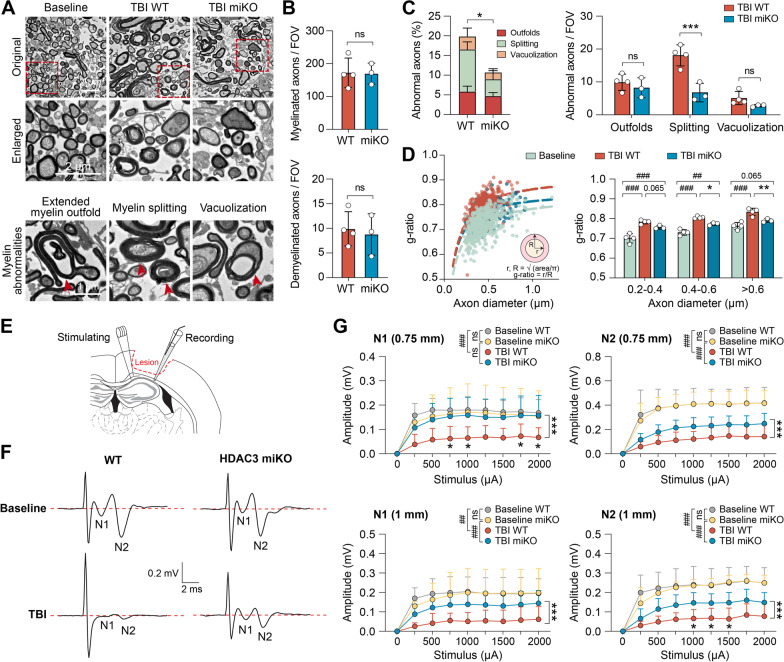
Methods: We generated HDAC3LoxP mice and crossed them with CX3CR1CreER mice, enabling in vivo conditional deletion of HDAC3. Microglia-specific HDAC3 knockout (HDAC3 miKO) was induced in CX3CR1CreER:HDAC3LoxP mice with 5 days of tamoxifen treatment followed by a 30-day development interval. The effects of HDAC3 miKO on microglial phenotype and neuroinflammation were examined 3-5 days after TBI induced by controlled cortical impact. Neurological deficits and the integrity of white matter were assessed for 6 weeks after TBI by neurobehavioral tests, immunohistochemistry, electron microscopy, and electrophysiology.
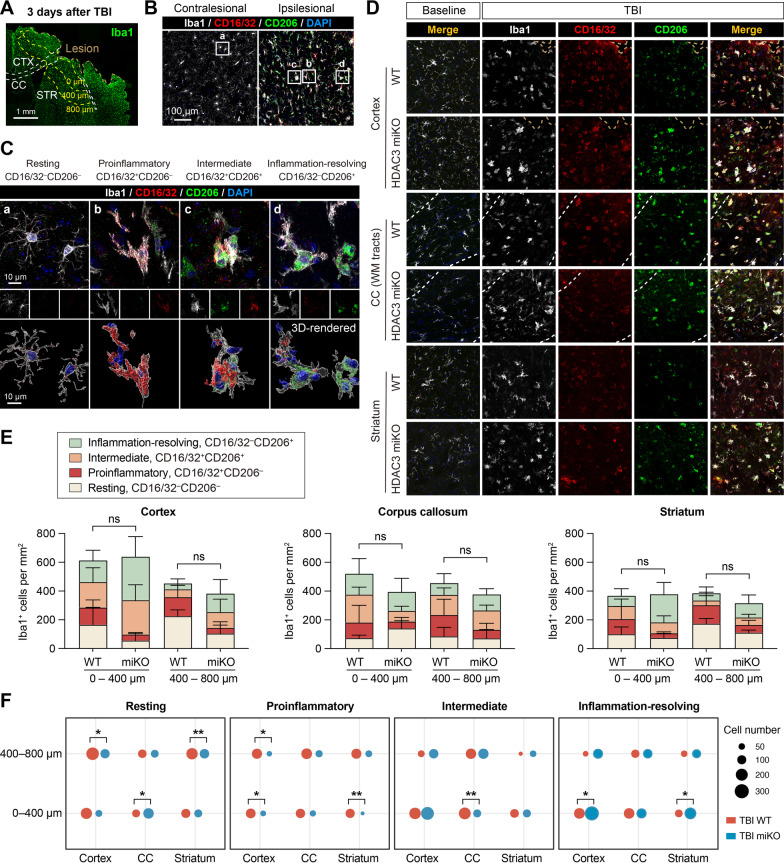
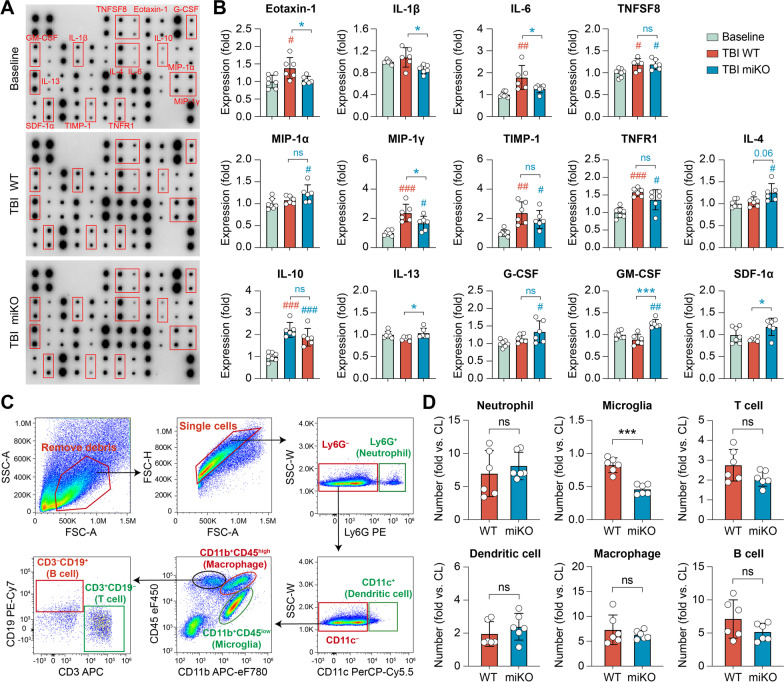
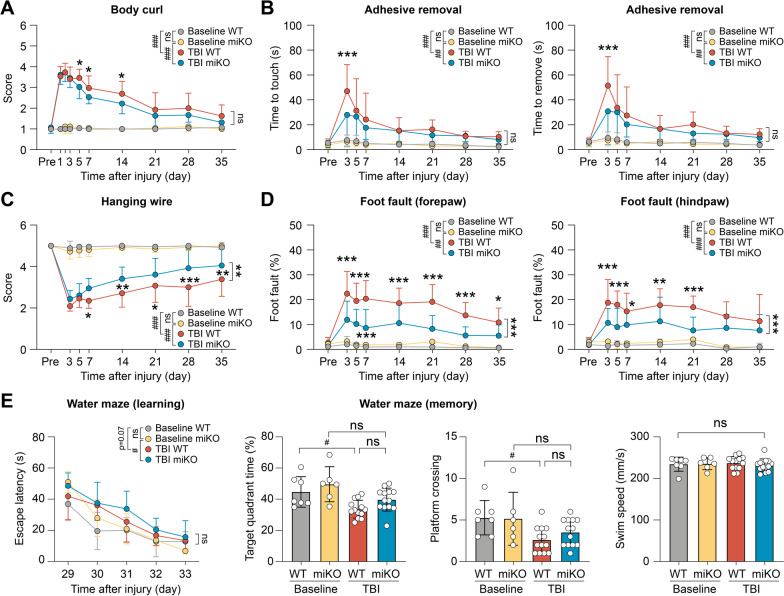
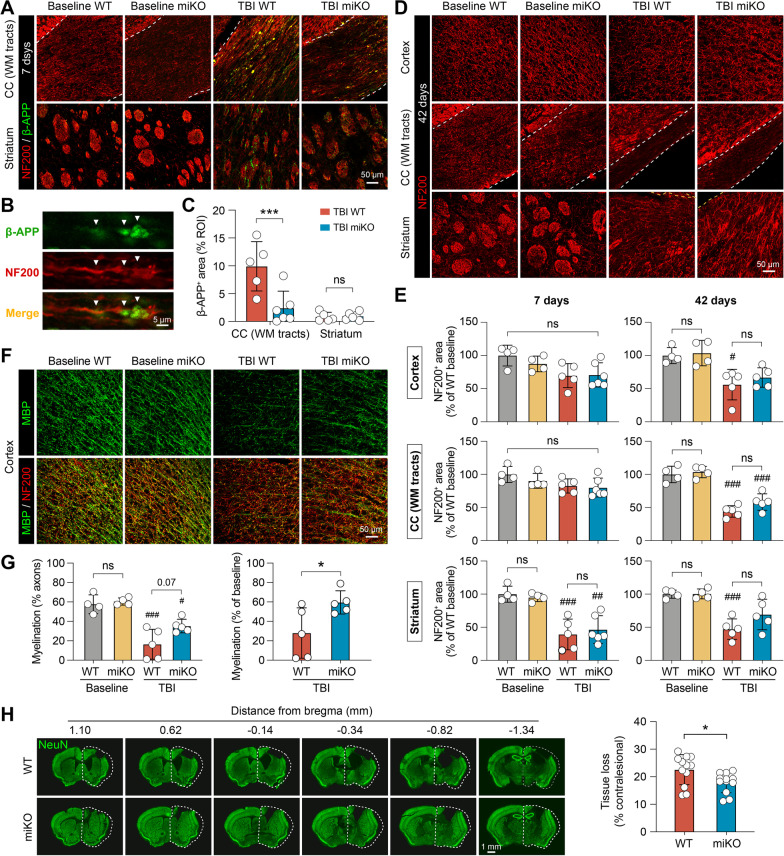
Results: HDAC3 miKO mice harbored specific deletion of HDAC3 in microglia but not in peripheral monocytes. HDAC3 miKO reduced the number of microglia by 26%, but did not alter the inflammation level in the homeostatic brain. After TBI, proinflammatory microglial responses and brain inflammation were markedly alleviated by HDAC3 miKO, whereas the infiltration of blood immune cells was unchanged, suggesting a primary effect of HDAC3 miKO on modulating microglial phenotype. Importantly, HDAC3 miKO was sufficient to facilitate functional recovery for 6 weeks after TBI. TBI-induced injury to axons and myelin was ameliorated, and signal conduction by white matter fiber tracts was significantly enhanced in HDAC3 miKO mice.
Conclusion: Using a novel microglia-specific conditional knockout mouse model, we delineated for the first time the role of microglial HDAC3 after TBI in vivo. HDAC3 miKO not only reduced proinflammatory microglial responses, but also elicited long-lasting improvement of white matter integrity and functional recovery after TBI. Microglial HDAC3 is therefore a promising therapeutic target to improve long-term outcomes after TBI.
Keywords: Conditional gene knockout; Controlled cortical impact; HDAC3; Neuroinflammation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Shein NA, Grigoriadis N, Alexandrovich AG, Simeonidou C, Lourbopoulos A, Polyzoidou E, et al. Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 2009;23(12):4266–4275. doi: 10.1096/fj.09-134700. - DOI - PMC - PubMed
-
- Wang G, Jiang X, Pu H, Zhang W, An C, Hu X, et al. Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway: scriptaid protects against TBI via AKT. Neurotherapeutics. 2013;10(1):124–142. doi: 10.1007/s13311-012-0157-2. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
