

Simultaneous testing of rule- and model-based approaches for runs of homozygosity detection opens up a window into genomic footprints of selection in pigs
- PMID: 35933356
- PMCID: PMC9357325
- DOI: 10.1186/s12864-022-08801-4
Simultaneous testing of rule- and model-based approaches for runs of homozygosity detection opens up a window into genomic footprints of selection in pigs
Abstract
Background: Past selection events left footprints in the genome of domestic animals, which can be traced back by stretches of homozygous genotypes, designated as runs of homozygosity (ROHs). The analysis of common ROH regions within groups or populations displaying potential signatures of selection requires high-quality SNP data as well as carefully adjusted ROH-defining parameters. In this study, we used a simultaneous testing of rule- and model-based approaches to perform strategic ROH calling in genomic data from different pig populations to detect genomic regions under selection for specific phenotypes.
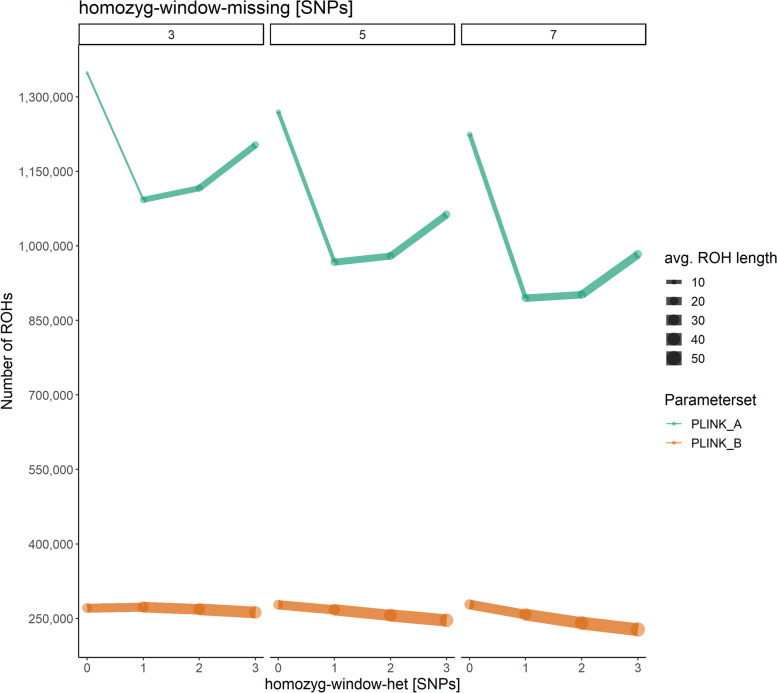
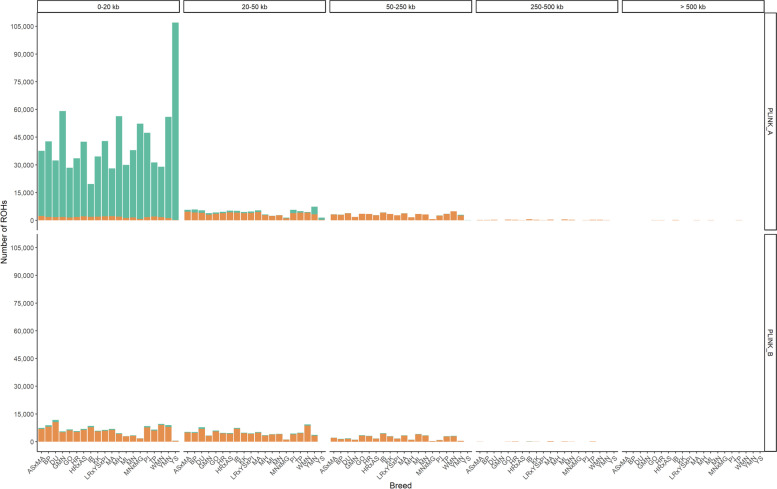
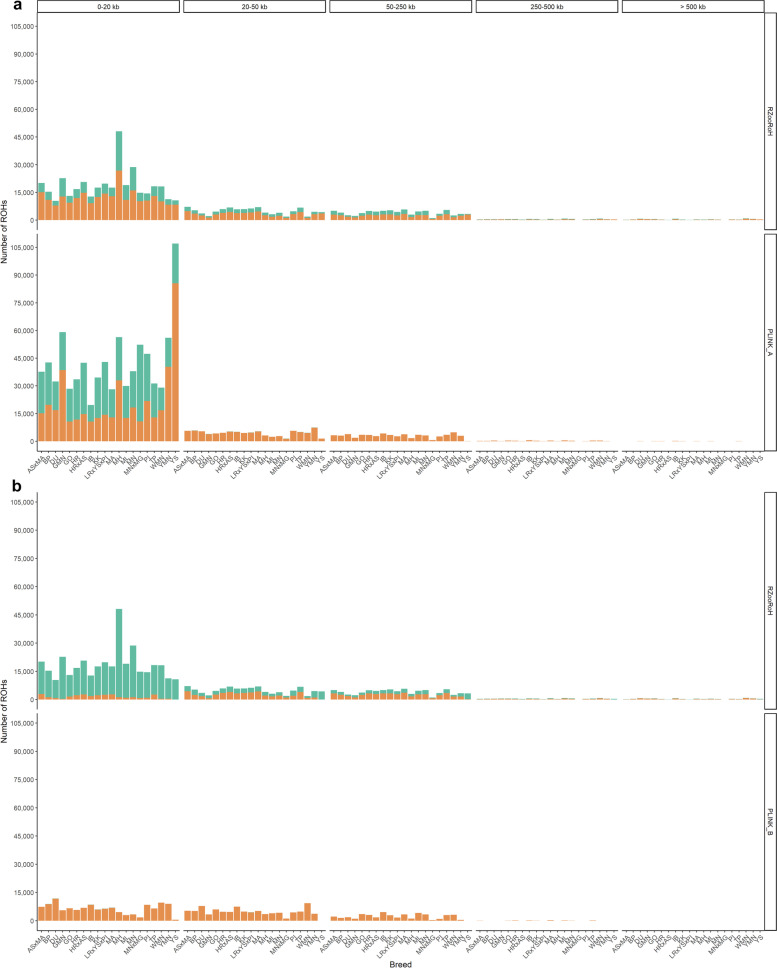
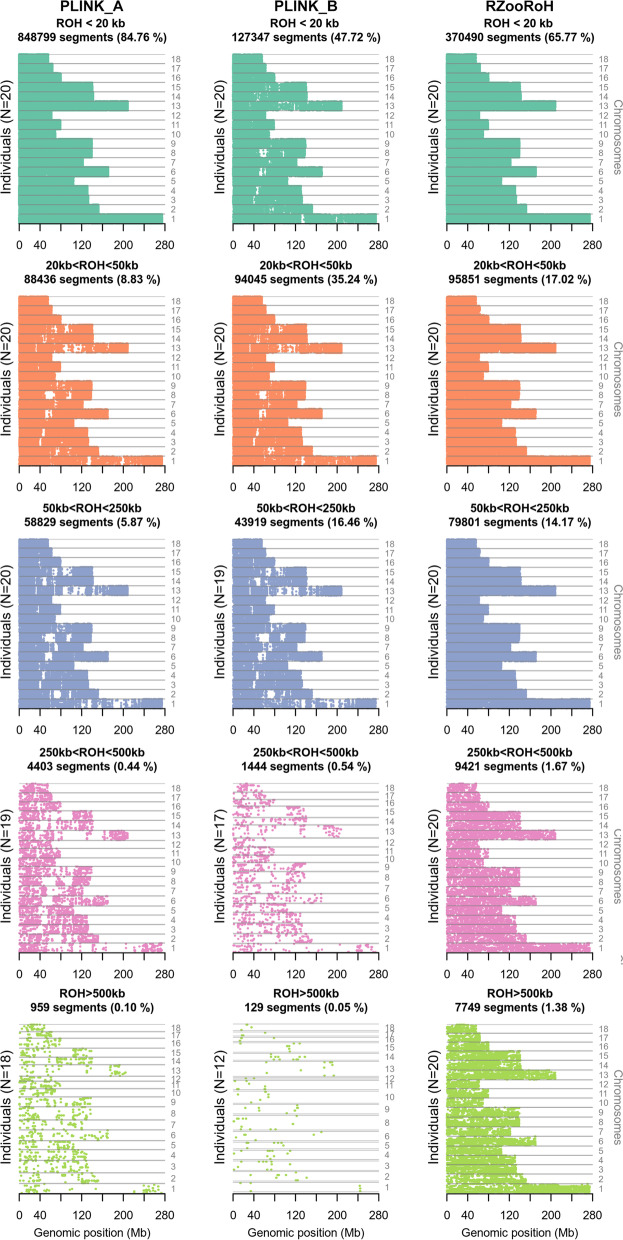
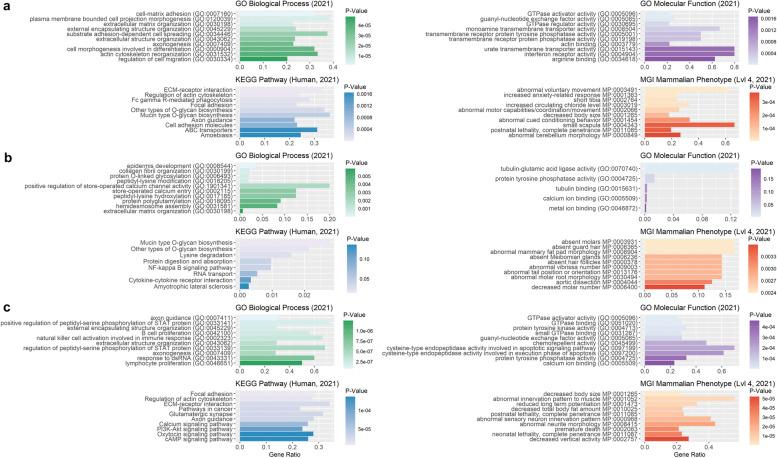
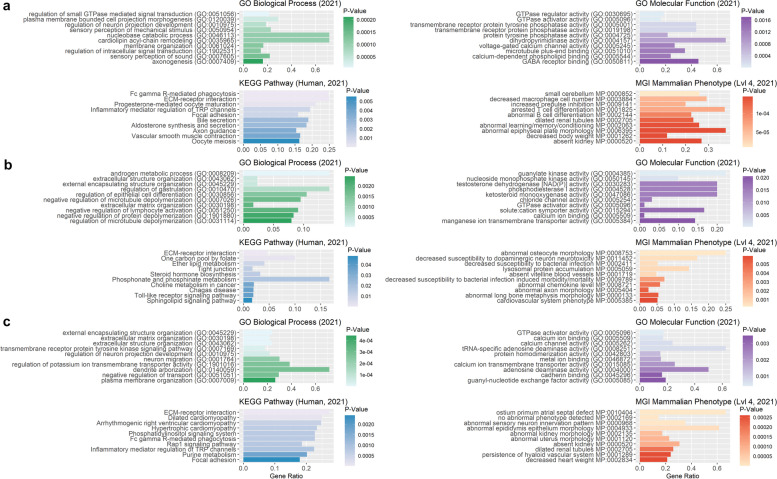
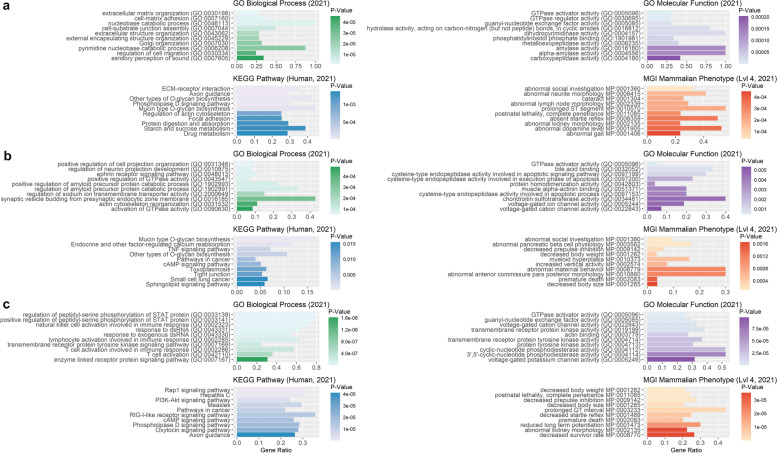
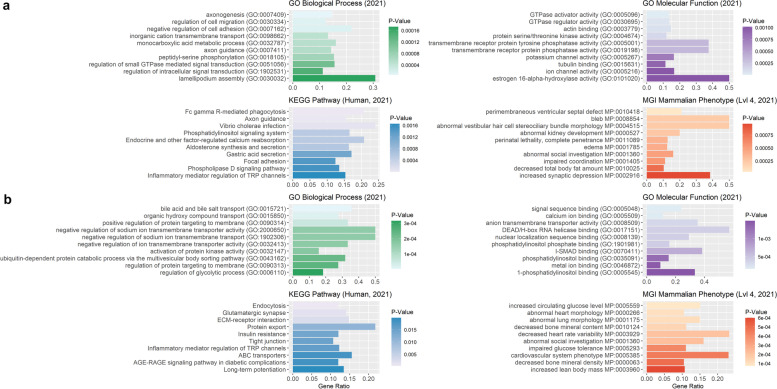
Results: Our ROH analysis using a rule-based approach offered by PLINK, as well as a model-based approach run by RZooRoH demonstrated a high efficiency of both methods. It underlined the importance of providing a high-quality SNP set as input as well as adjusting parameters based on dataset and population for ROH calling. Particularly, ROHs ≤ 20 kb were called in a high frequency by both tools, but to some extent covered different gene sets in subsequent analysis of ROH regions common for investigated pig groups. Phenotype associated ROH analysis resulted in regions under potential selection characterizing heritage pig breeds, known to harbour a long-established breeding history. In particular, the selection focus on fitness-related traits was underlined by various ROHs harbouring disease resistance or tolerance-associated genes. Moreover, we identified potential selection signatures associated with ear morphology, which confirmed known candidate genes as well as uncovered a missense mutation in the ABCA6 gene potentially supporting ear cartilage formation.
Conclusions: The results of this study highlight the strengths and unique features of rule- and model-based approaches as well as demonstrate their potential for ROH analysis in animal populations. We provide a workflow for ROH detection, evaluating the major steps from filtering for high-quality SNP sets to intersecting ROH regions. Formula-based estimations defining ROHs for rule-based method show its limits, particularly for efficient detection of smaller ROHs. Moreover, we emphasize the role of ROH detection for the identification of potential footprints of selection in pigs, displaying their breed-specific characteristics or favourable phenotypes.
Keywords: Pig genome; Runs of homozygosity; SNP density; Selection signatures.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
Runs of homozygosity provide a genome landscape picture of inbreeding and genetic history of European autochthonous and commercial pig breeds.Anim Genet. 2021 Apr;52(2):155-170. doi: 10.1111/age.13045. Epub 2021 Feb 5. Anim Genet. 2021. PMID: 33544919
-
Characterization of runs of Homozygosity revealed genomic inbreeding and patterns of selection in indigenous sahiwal cattle.J Appl Genet. 2024 Feb;65(1):167-180. doi: 10.1007/s13353-023-00816-1. Epub 2023 Dec 18. J Appl Genet. 2024. PMID: 38110827
-
Runs of homozygosity reveal signatures of positive selection for reproduction traits in breed and non-breed horses.BMC Genomics. 2015 Oct 9;16:764. doi: 10.1186/s12864-015-1977-3. BMC Genomics. 2015. PMID: 26452642 Free PMC article.
-
Genome-wide selection signatures detection in Shanghai Holstein cattle population identified genes related to adaption, health and reproduction traits.BMC Genomics. 2021 Oct 15;22(1):747. doi: 10.1186/s12864-021-08042-x. BMC Genomics. 2021. PMID: 34654366 Free PMC article. Review.
-
Runs of homozygosity: current knowledge and applications in livestock.Anim Genet. 2017 Jun;48(3):255-271. doi: 10.1111/age.12526. Epub 2016 Dec 1. Anim Genet. 2017. PMID: 27910110 Review.
Cited by
-
Genetic diversity and reproductive trait selection signal analysis of Bohuai goats based on whole genome sequencing.BMC Genomics. 2025 Jul 26;26(1):693. doi: 10.1186/s12864-025-11878-2. BMC Genomics. 2025. PMID: 40713489 Free PMC article.
-
Performance of homozygosity by descent in two mice lines divergently selected for birth weight environmental variability.Sci Rep. 2025 Feb 14;15(1):5511. doi: 10.1038/s41598-025-89254-z. Sci Rep. 2025. PMID: 39953099 Free PMC article.
-
Multi-tool copy number detection highlights common body size-associated variants in miniature pig breeds from different geographical regions.BMC Genomics. 2025 Mar 22;26(1):285. doi: 10.1186/s12864-025-11446-8. BMC Genomics. 2025. PMID: 40121435 Free PMC article.
References
-
- Falconer DS, Mackay TFC. Introduction to quantitative genetics. 4. Harlow, England: Prentice Hall; 1996.
-
- Curik I, Ferenčaković M, Sölkner J. Inbreeding and runs of homozygosity: a possible solution to an old problem. Livest Sci. 2014;166:26–34. doi: 10.1016/j.livsci.2014.05.034. - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
