

Three Newly Recognized Likely Pathogenic Gene Variants Associated with Hereditary Transthyretin Amyloidosis
- PMID: 35933469
- PMCID: PMC9588125
- DOI: 10.1007/s40120-022-00385-1
Three Newly Recognized Likely Pathogenic Gene Variants Associated with Hereditary Transthyretin Amyloidosis
Abstract
Introduction: Hereditary transthyretin amyloidosis (ATTRv [variant]) is a clinically heterogeneous, progressively debilitating, fatal disease resulting from the deposition of insoluble amyloid fibrils in various organs and tissues. Early diagnosis of ATTRv can be facilitated with genetic testing; however, such testing of the TTR gene identifies variants of uncertain significance (VUS) in a minority of cases, a small percentage of which have the potential to be pathogenic. The Akcea/Ambry VUS Initiative is dedicated to gathering molecular, clinical, and inheritance data for each TTR VUS identified by genetic testing programs to reclassify TTR variants to a clinically actionable status (e.g., variant likely pathogenic [VLP]) where appropriate.
Methods: Classification criteria used here, based on recommendations from the American College of Medical Genetics and Genomics, are stringent and comprehensive, requiring distinct lines of evidence supporting pathogenesis.
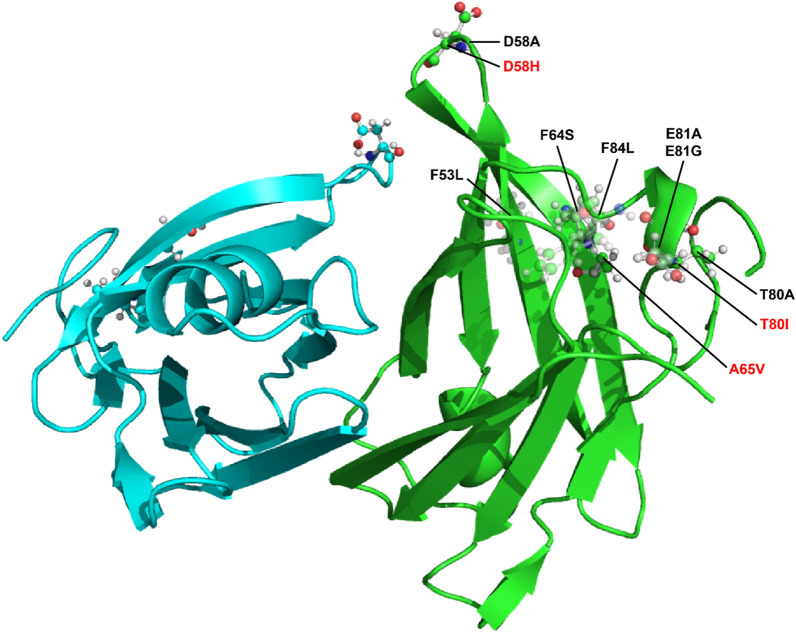
Results: Three TTR variants have been reclassified from VUS to VLP, including c.194C>T (p.A65V), c.172G>C (p.D58H), and c.239C>T (p.T80I). In each case, the totality of genetic, structural, and clinical evidence provided strong support for pathogenicity.
Conclusions: Based on several lines of evidence, three TTR VUS were reclassified as VLP, resulting in a high likelihood of disease diagnosis for those and subsequent patients as well as at-risk family members.
Keywords: Hereditary transthyretin amyloidosis; Likely pathogenic variant; Reclassification, variant of uncertain significance; Variant of unknown significance.
© 2022. The Author(s).
Figures
References
-
- Sekijima Y. Hereditary transthyretin amyloidosis. Gene Reviews. Seattle: University of Washington; 2018.
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
