

Multi-omics personalized network analyses highlight progressive disruption of central metabolism associated with COVID-19 severity
- PMID: 35933992
- PMCID: PMC9263811
- DOI: 10.1016/j.cels.2022.06.006
Multi-omics personalized network analyses highlight progressive disruption of central metabolism associated with COVID-19 severity
Abstract
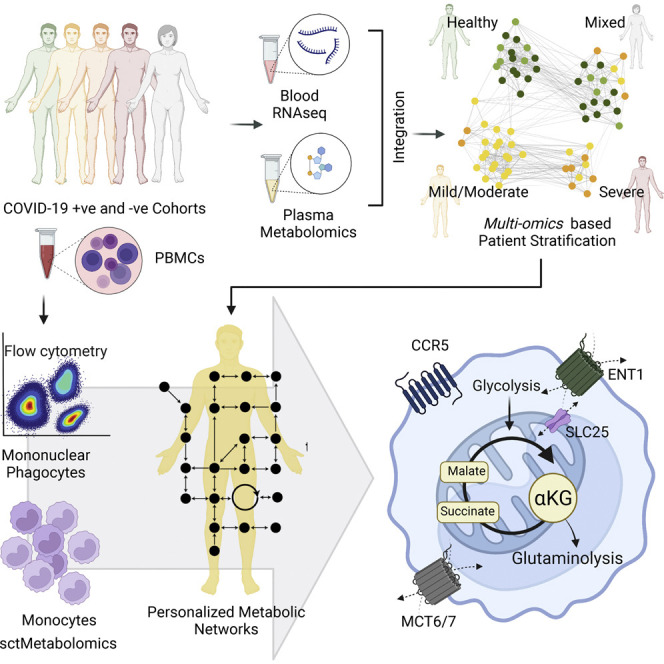
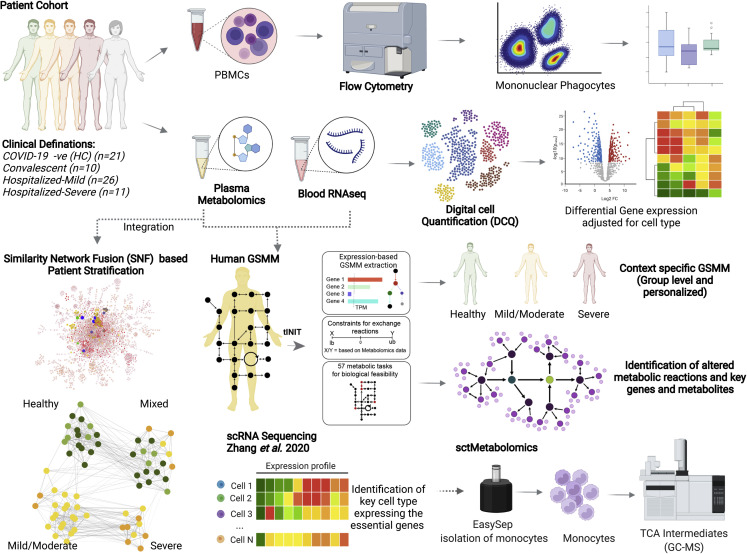
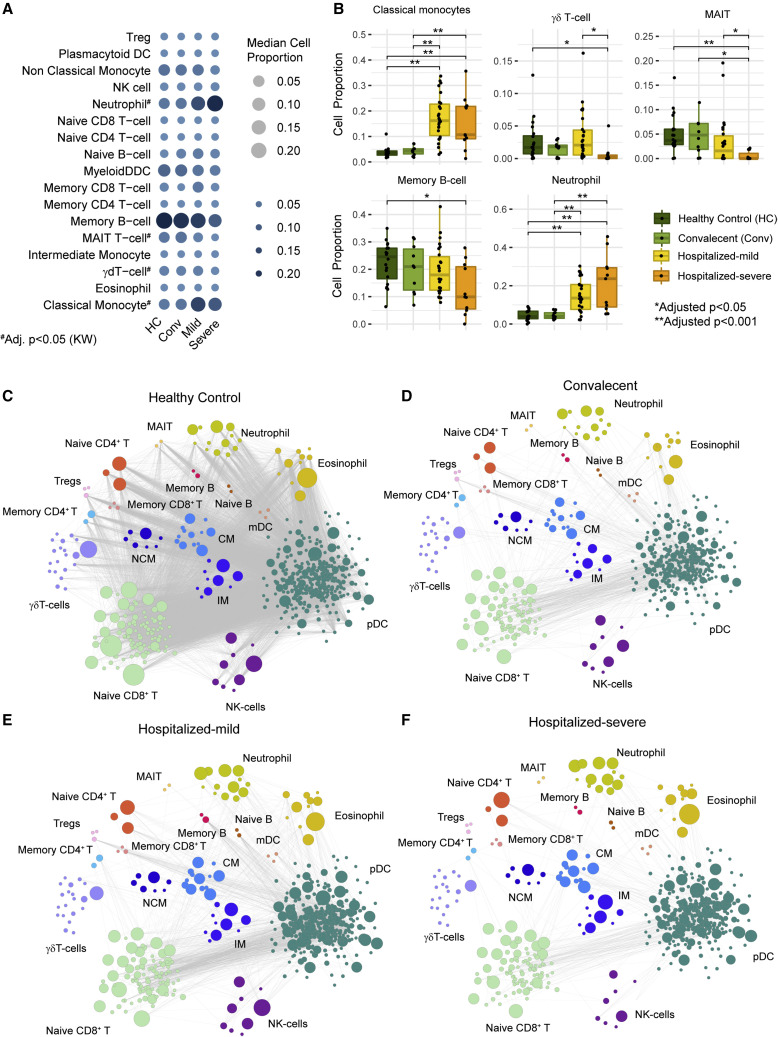
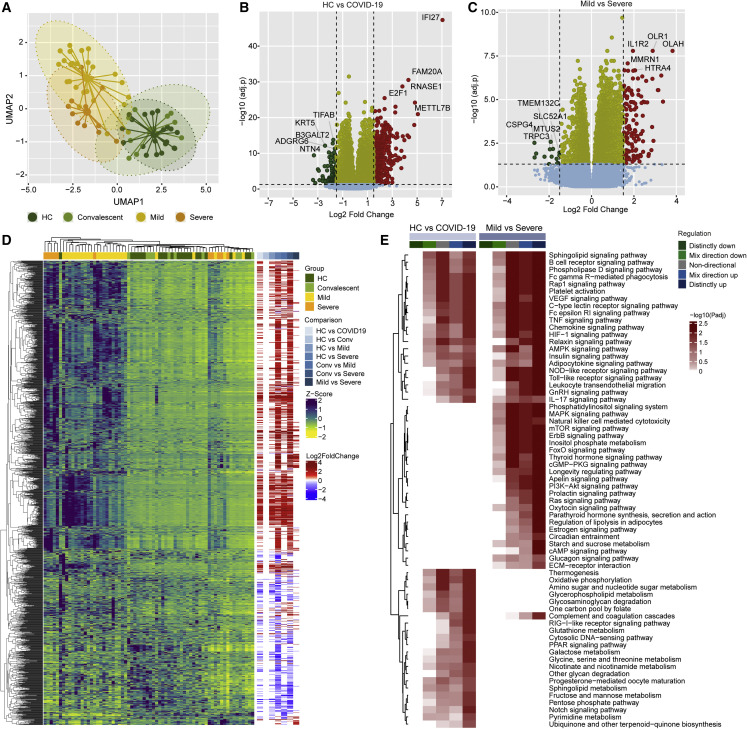
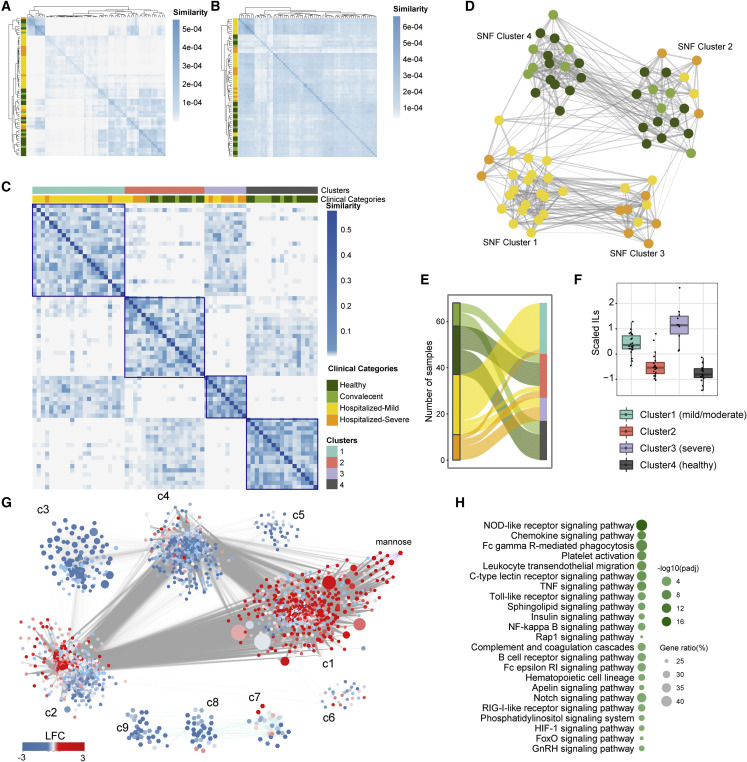
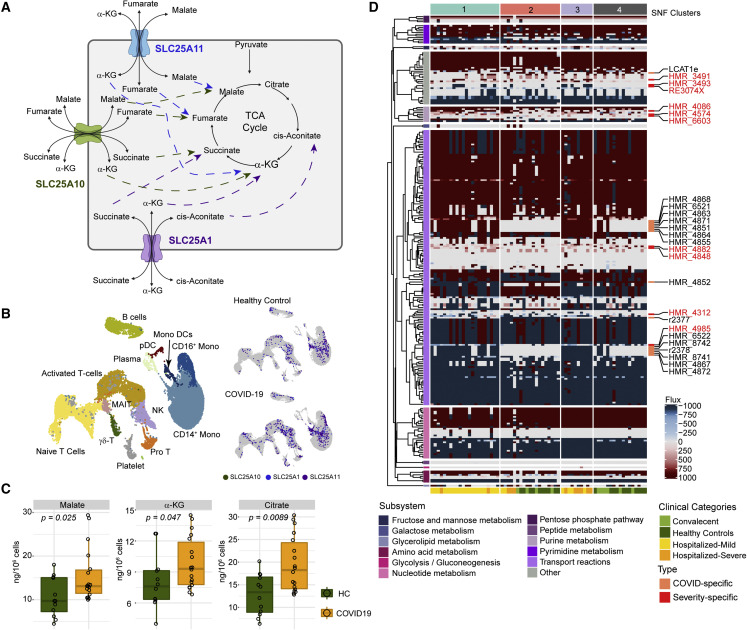
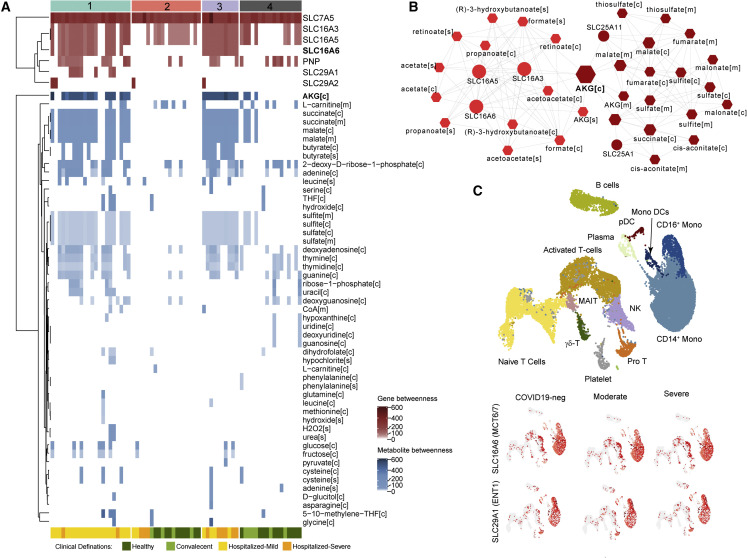
The clinical outcome and disease severity in coronavirus disease 2019 (COVID-19) are heterogeneous, and the progression or fatality of the disease cannot be explained by a single factor like age or comorbidities. In this study, we used system-wide network-based system biology analysis using whole blood RNA sequencing, immunophenotyping by flow cytometry, plasma metabolomics, and single-cell-type metabolomics of monocytes to identify the potential determinants of COVID-19 severity at personalized and group levels. Digital cell quantification and immunophenotyping of the mononuclear phagocytes indicated a substantial role in coordinating the immune cells that mediate COVID-19 severity. Stratum-specific and personalized genome-scale metabolic modeling indicated monocarboxylate transporter family genes (e.g., SLC16A6), nucleoside transporter genes (e.g., SLC29A1), and metabolites such as α-ketoglutarate, succinate, malate, and butyrate could play a crucial role in COVID-19 severity. Metabolic perturbations targeting the central metabolic pathway (TCA cycle) can be an alternate treatment strategy in severe COVID-19.
Keywords: COVID-19; personalized genome-scale metabolic model; similarity network fusion.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures

References
-
- Albrich W.C., Ghosh T.S., Ahearn-Ford S., Mikaeloff F., Lunjani N., Forde B., Suh N., Kleger G.R., Pietsch U., Frischknecht M., et al. A high-risk gut microbiota configuration associates with fatal hyperinflammatory immune and metabolic responses to SARS-CoV-2. Gut Microbes. 2022;14:2073131. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
