

Drug development progress in duchenne muscular dystrophy
- PMID: 35935842
- PMCID: PMC9353054
- DOI: 10.3389/fphar.2022.950651
Drug development progress in duchenne muscular dystrophy
Abstract
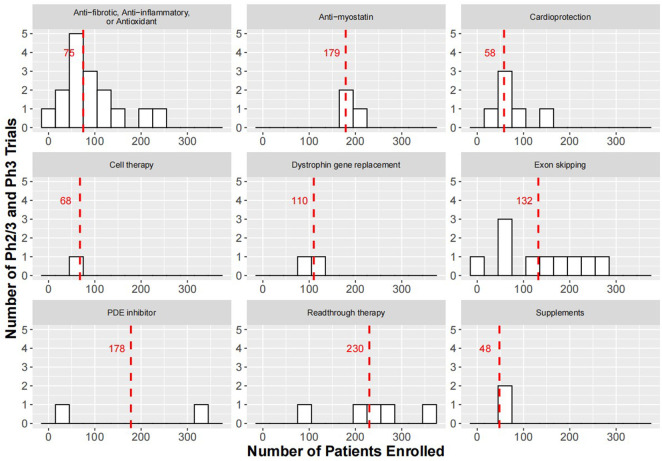
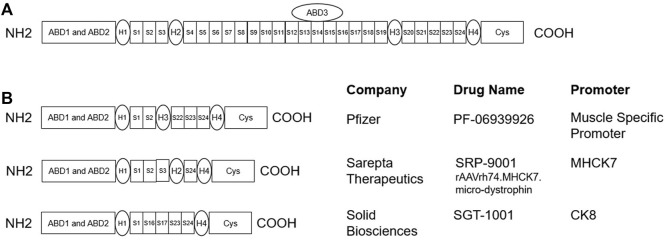
Duchenne muscular dystrophy (DMD) is a severe, progressive, and incurable X-linked disorder caused by mutations in the dystrophin gene. Patients with DMD have an absence of functional dystrophin protein, which results in chronic damage of muscle fibers during contraction, thus leading to deterioration of muscle quality and loss of muscle mass over time. Although there is currently no cure for DMD, improvements in treatment care and management could delay disease progression and improve quality of life, thereby prolonging life expectancy for these patients. Furthermore, active research efforts are ongoing to develop therapeutic strategies that target dystrophin deficiency, such as gene replacement therapies, exon skipping, and readthrough therapy, as well as strategies that target secondary pathology of DMD, such as novel anti-inflammatory compounds, myostatin inhibitors, and cardioprotective compounds. Furthermore, longitudinal modeling approaches have been used to characterize the progression of MRI and functional endpoints for predictive purposes to inform Go/No Go decisions in drug development. This review showcases approved drugs or drug candidates along their development paths and also provides information on primary endpoints and enrollment size of Ph2/3 and Ph3 trials in the DMD space.
Keywords: clinical trial; drug developement; duchenne muscular dystrophy (DMD); research and development; therapeutic strategies.
Copyright © 2022 Deng, Zhang, Shi and Liu.
Conflict of interest statement
JD was employed by the company, Pfizer Inc. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Molecular and Biochemical Therapeutic Strategies for Duchenne Muscular Dystrophy.Neurol Int. 2024 Jul 5;16(4):731-760. doi: 10.3390/neurolint16040055. Neurol Int. 2024. PMID: 39051216 Free PMC article. Review.
-
Causes of clinical variability in Duchenne and Becker muscular dystrophies and implications for exon skipping therapies.Acta Myol. 2020 Dec 1;39(4):179-186. doi: 10.36185/2532-1900-020. eCollection 2020 Dec. Acta Myol. 2020. PMID: 33458572 Free PMC article.
-
Mutation-Based Therapeutic Strategies for Duchenne Muscular Dystrophy: From Genetic Diagnosis to Therapy.J Pers Med. 2019 Mar 4;9(1):16. doi: 10.3390/jpm9010016. J Pers Med. 2019. PMID: 30836656 Free PMC article. Review.
-
Developing DMD therapeutics: a review of the effectiveness of small molecules, stop-codon readthrough, dystrophin gene replacement, and exon-skipping therapies.Expert Opin Investig Drugs. 2021 Feb;30(2):167-176. doi: 10.1080/13543784.2021.1868434. Epub 2021 Jan 6. Expert Opin Investig Drugs. 2021. PMID: 33393390 Review.
-
De novo revertant fiber formation and therapy testing in a 3D culture model of Duchenne muscular dystrophy skeletal muscle.Acta Biomater. 2021 Sep 15;132:227-244. doi: 10.1016/j.actbio.2021.05.020. Epub 2021 May 25. Acta Biomater. 2021. PMID: 34048976
Cited by
-
Safety and Efficacy of DT-DEC01 Therapy in Duchenne Muscular Dystrophy Patients: A 12 - Month Follow-Up Study After Systemic Intraosseous Administration.Stem Cell Rev Rep. 2023 Nov;19(8):2724-2740. doi: 10.1007/s12015-023-10620-3. Epub 2023 Sep 14. Stem Cell Rev Rep. 2023. PMID: 37707670 Free PMC article.
-
Mechanisms and effects of metformin on skeletal muscle disorders.Front Neurol. 2023 Oct 19;14:1275266. doi: 10.3389/fneur.2023.1275266. eCollection 2023. Front Neurol. 2023. PMID: 37928155 Free PMC article. Review.
-
Unveiling the Future of Cardiac Care: A Review of Gene Therapy in Cardiomyopathies.Int J Mol Sci. 2024 Dec 6;25(23):13147. doi: 10.3390/ijms252313147. Int J Mol Sci. 2024. PMID: 39684857 Free PMC article. Review.
-
Uncovering the Embryonic Origins of Duchenne Muscular Dystrophy.WIREs Mech Dis. 2024 Nov-Dec;16(6):e1653. doi: 10.1002/wsbm.1653. Epub 2024 Oct 23. WIREs Mech Dis. 2024. PMID: 39444092
-
Have a Little Heart (or Not): Highly Minimized Skeletal Muscle Regulatory Cassettes with Low or No Activity in the Heart.Hum Gene Ther. 2024 Aug;35(15-16):543-554. doi: 10.1089/hum.2024.041. Epub 2024 Jul 19. Hum Gene Ther. 2024. PMID: 38970421 Free PMC article.
References
-
- Aartsma-Rus A., Straub V., Hemmings R., Haas M., Schlosser-Weber G., Stoyanova-Beninska V., et al. (2017). Development of exon skipping therapies for duchenne muscular dystrophy: A critical review and a perspective on the outstanding issues. Nucleic Acid. Ther. 27 (5), 251–259. 10.1089/nat.2017.0682 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources