

A probable cis-acting genetic modifier of Huntington disease frequent in individuals with African ancestry
- PMID: 35935919
- PMCID: PMC9352962
- DOI: 10.1016/j.xhgg.2022.100130
A probable cis-acting genetic modifier of Huntington disease frequent in individuals with African ancestry
Abstract
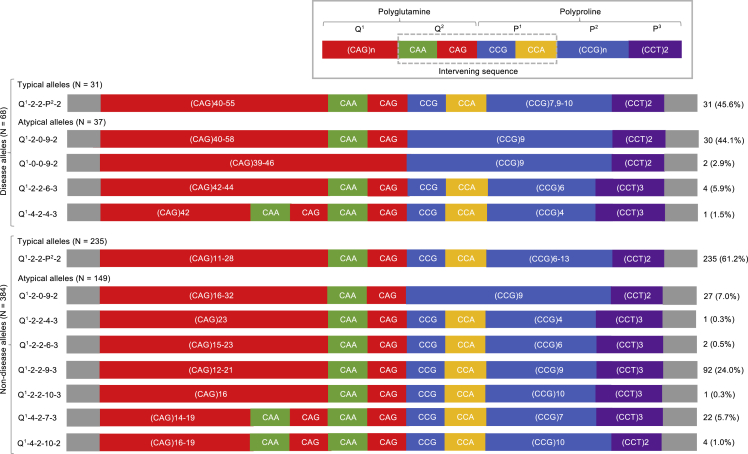
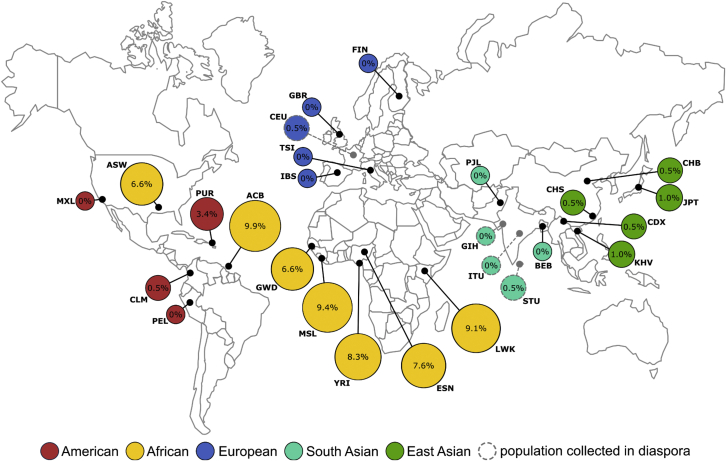
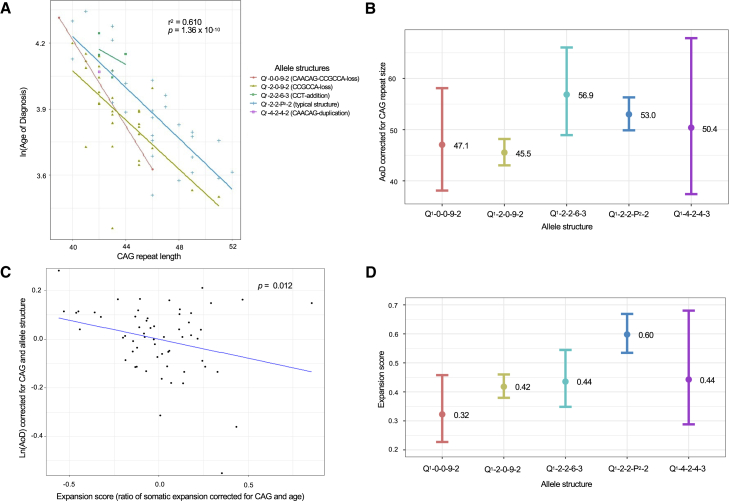
Huntington disease (HD)is a dominantly inherited neurodegenerative disorder caused by the expansion of a polyglutamine encoding CAG repeat in the huntingtin gene. Recently, it has been established that disease severity in HD is best predicted by the number of pure CAG repeats rather than total glutamines encoded. Along with uncovering DNA repair gene variants as trans-acting modifiers of HD severity, these data reveal somatic expansion of the CAG repeat as a key driver of HD onset. Using high-throughput DNA sequencing, we have determined the precise sequence and somatic expansion profiles of the HTT repeat tract of 68 HD-affected and 158 HD-unaffected African ancestry individuals. A high level of HTT repeat sequence diversity was observed, with three likely African-specific alleles identified. In the most common disease allele (30 out of 68), the typical proline-encoding CCGCCA sequence was absent. This CCGCCA-loss disease allele was associated with an earlier age of diagnosis of approximately 7.1 years and occurred exclusively on haplotype B2. Although somatic expansion was associated with an earlier age of diagnosis in the study overall, the CCGCCA-loss disease allele displayed reduced somatic expansion relative to the typical HTT expansions in blood DNA. We propose that the CCGCCA loss occurring on haplotype B2 is an African cis-acting modifier that appears to alter disease diagnosis of HD through a mechanism that is not driven by somatic expansion. The assessment of a group of individuals from an understudied population has highlighted population-specific differences that emphasize the importance of studying genetically diverse populations in the context of disease.
Keywords: African ancestry; CAG repeat; CCGCCA loss; Huntington disease; cis-acting modifier; genetically diverse.
© 2022 The Author(s).
Conflict of interest statement
Within the last 5 years, D.G.M. has been a scientific consultant and/or received an honoraria/stock options/grants from AMO Pharma, Charles River, LoQus23, Small Molecule RNA, Triplet Therapeutics, and Vertex Pharmaceuticals. D.G.M. also had research contracts with AMO Pharma and Vertex Pharmaceuticals. The other authors declare no competing interests.
Figures
Similar articles
-
A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes.EBioMedicine. 2019 Oct;48:568-580. doi: 10.1016/j.ebiom.2019.09.020. Epub 2019 Oct 10. EBioMedicine. 2019. PMID: 31607598 Free PMC article.
-
Length of Uninterrupted CAG, Independent of Polyglutamine Size, Results in Increased Somatic Instability, Hastening Onset of Huntington Disease.Am J Hum Genet. 2019 Jun 6;104(6):1116-1126. doi: 10.1016/j.ajhg.2019.04.007. Epub 2019 May 16. Am J Hum Genet. 2019. PMID: 31104771 Free PMC article.
-
Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington's disease knock-in mice is blocked by Mlh1 knock-out.Hum Mol Genet. 2020 Nov 4;29(18):3044-3053. doi: 10.1093/hmg/ddaa196. Hum Mol Genet. 2020. PMID: 32876667 Free PMC article.
-
Beyond CAG Repeats: The Multifaceted Role of Genetics in Huntington Disease.Genes (Basel). 2024 Jun 19;15(6):807. doi: 10.3390/genes15060807. Genes (Basel). 2024. PMID: 38927742 Free PMC article. Review.
-
Modifiers of Somatic Repeat Instability in Mouse Models of Friedreich Ataxia and the Fragile X-Related Disorders: Implications for the Mechanism of Somatic Expansion in Huntington's Disease.J Huntingtons Dis. 2021;10(1):149-163. doi: 10.3233/JHD-200423. J Huntingtons Dis. 2021. PMID: 33579860 Free PMC article. Review.
Cited by
-
Spanish HTT gene study reveals haplotype and allelic diversity with possible implications for germline expansion dynamics in Huntington disease.Hum Mol Genet. 2023 Mar 6;32(6):897-906. doi: 10.1093/hmg/ddac224. Hum Mol Genet. 2023. PMID: 36130218 Free PMC article.
-
Genetic modifiers of repeat expansion disorders.Emerg Top Life Sci. 2023 Dec 14;7(3):325-337. doi: 10.1042/ETLS20230015. Emerg Top Life Sci. 2023. PMID: 37861103 Free PMC article.
-
Ascertainment of uninterrupted CAG repeat length and disease-modifying variants in fragment-based genetic testing for Huntington Disease.Genet Med Open. 2024 Aug 2;2:101882. doi: 10.1016/j.gimo.2024.101882. eCollection 2024. Genet Med Open. 2024. PMID: 39669608 Free PMC article.
-
Increased frequency of repeat expansion mutations across different populations.medRxiv [Preprint]. 2024 Jul 8:2023.07.03.23292162. doi: 10.1101/2023.07.03.23292162. medRxiv. 2024. Update in: Nat Med. 2024 Nov;30(11):3357-3368. doi: 10.1038/s41591-024-03190-5. PMID: 37461547 Free PMC article. Updated. Preprint.
-
Genetic modifiers of somatic expansion and clinical phenotypes in Huntington's disease highlight shared and tissue-specific effects.Nat Genet. 2025 Jun;57(6):1426-1436. doi: 10.1038/s41588-025-02191-5. Epub 2025 Jun 9. Nat Genet. 2025. PMID: 40490511
References
-
- The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on huntington’s disease chromosomes. Cell. 1993;72:971–983. - PubMed
-
- Walker F.O. Huntington's disease. Lancet. 2007;369:218–228. - PubMed
-
- Duyao M., Ambrose C., Myers R., Novelletto A., Persichetti F., Frontali M., Folstein S., Ross C., Franz M., Abbott M., et al. Trinucleotide repeat length instability and age of onset in huntington's disease. Nat. Genet. 1993;4:387–392. - PubMed
-
- Kennedy L., Evans E., Chen C.M., Craven L., Detloff P.J., Ennis M., Shelbourne P.F. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet. 2003;12:3359–3367. - PubMed
LinkOut - more resources
Full Text Sources
