

Silencing TUFM Inhibits Development of Monocrotaline-Induced Pulmonary Hypertension by Regulating Mitochondrial Autophagy via AMPK/mTOR Signal Pathway
- PMID: 35936222
- PMCID: PMC9348918
- DOI: 10.1155/2022/4931611
Silencing TUFM Inhibits Development of Monocrotaline-Induced Pulmonary Hypertension by Regulating Mitochondrial Autophagy via AMPK/mTOR Signal Pathway
Abstract
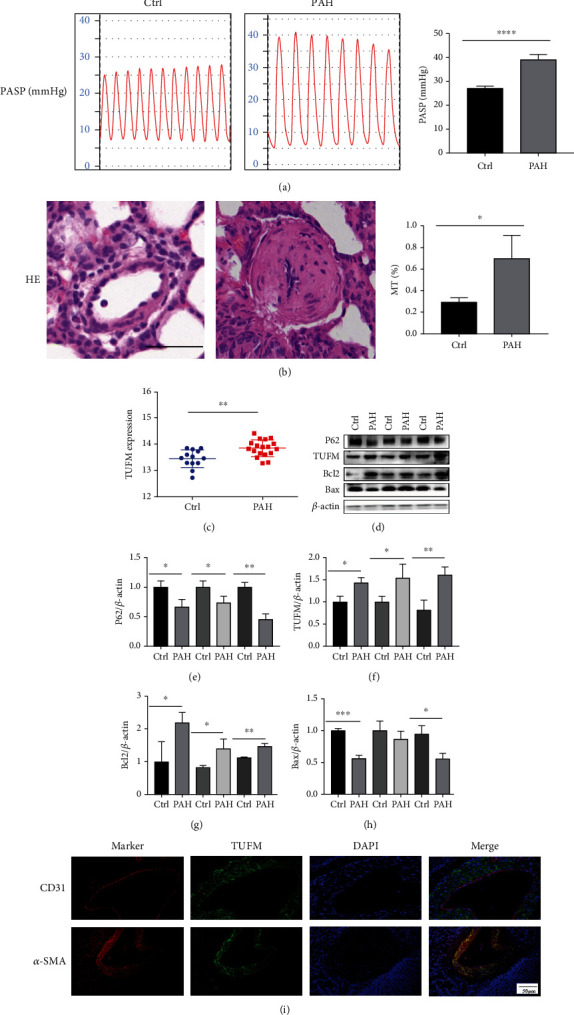
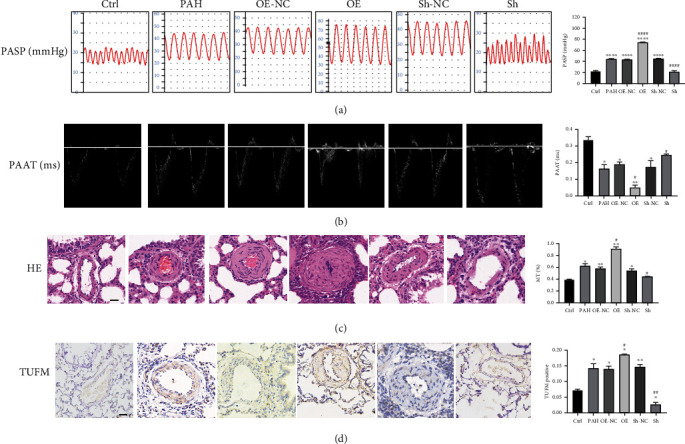
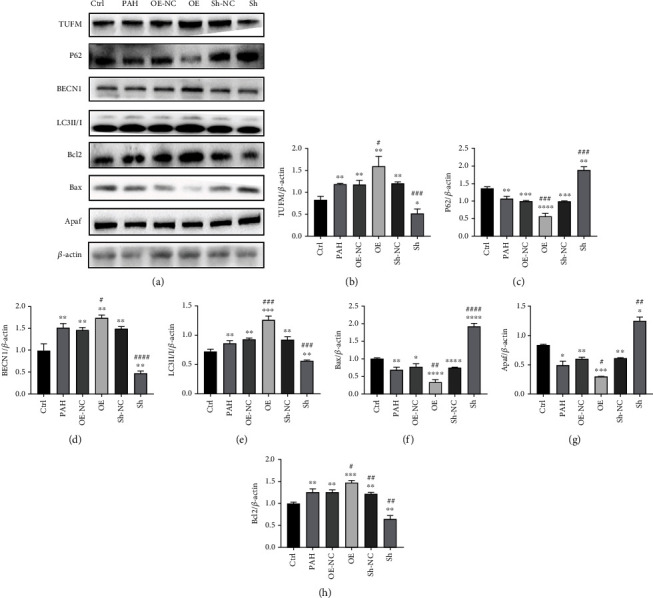
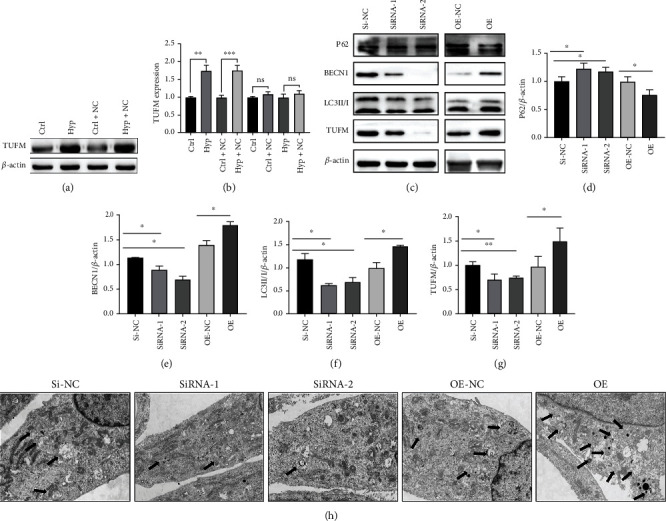
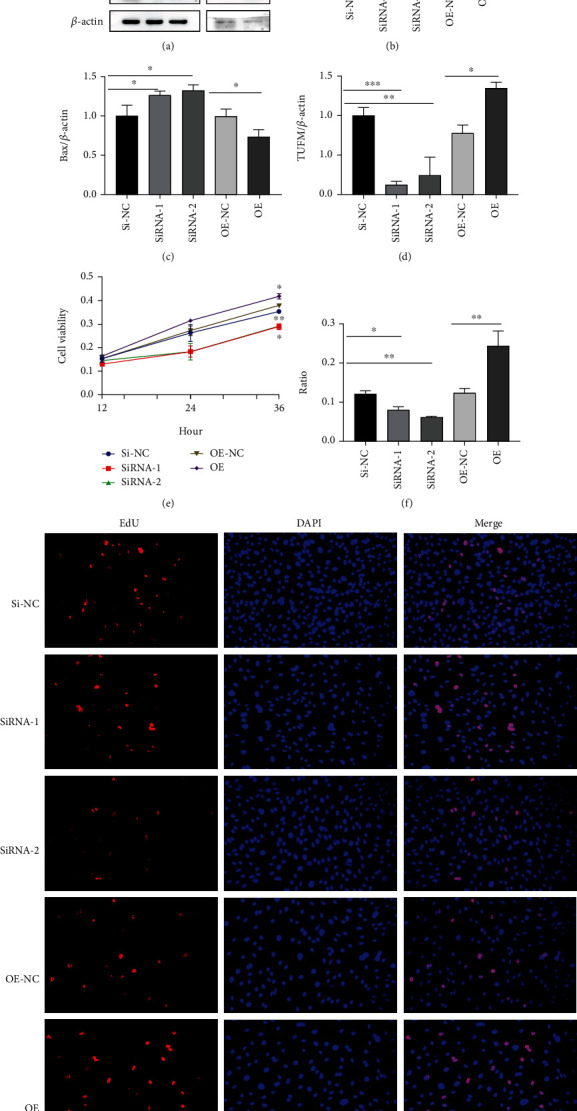
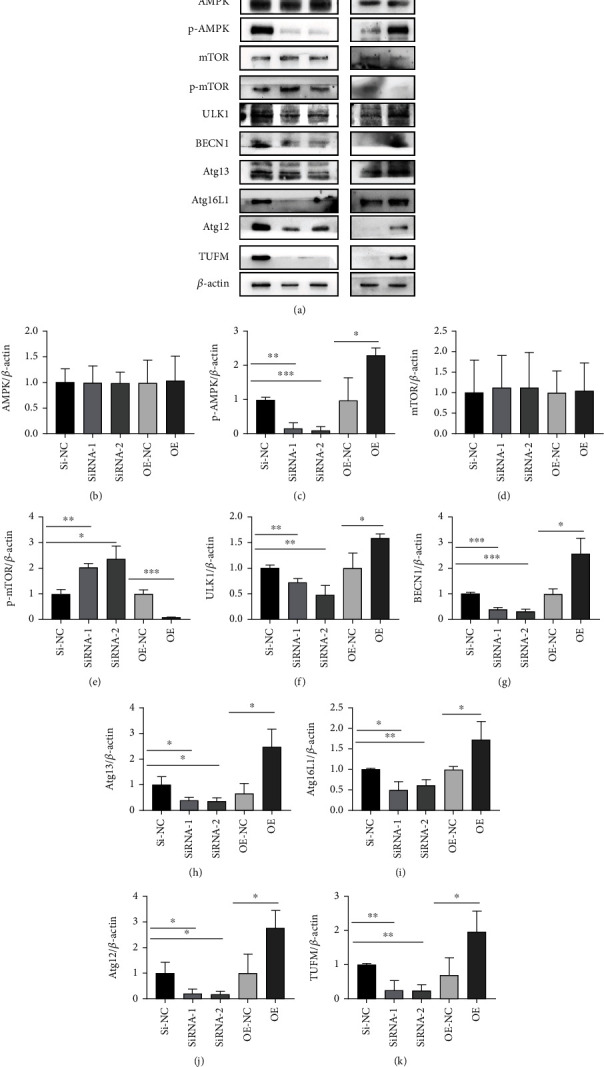
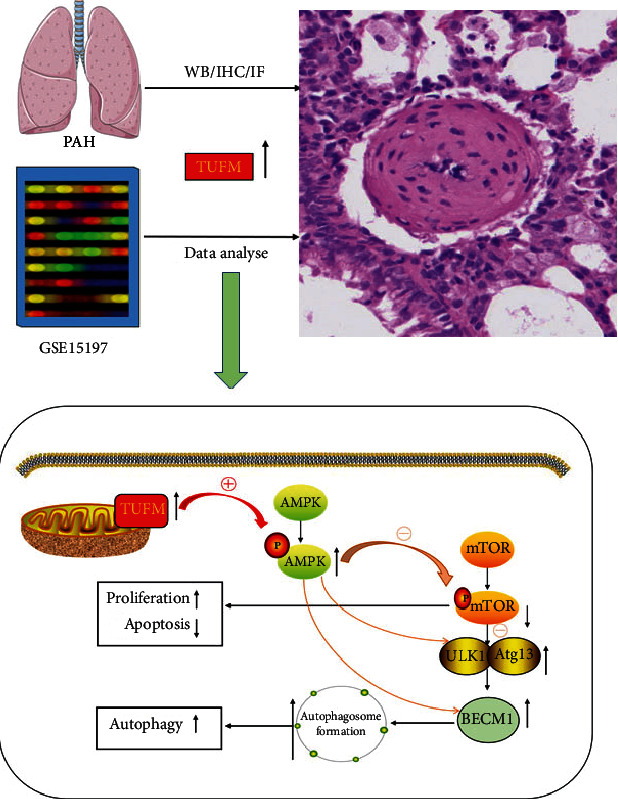
Pulmonary arterial hypertension (PAH) is an extremely malignant cardiovascular disease which mainly involves the uncontrollable proliferation of the pulmonary arterial smooth muscular cells (PASMCs). Recent studies have confirmed that mitochondria play an important role in the pathogenesis of pulmonary hypertension through sensing cell hypoxia, energy metabolism conversion, and apoptosis. As a mitochondrial membrane protein, TUFM has been regarded to be related to mitochondrial autophagy (mitophagy), apoptosis, and oxidative stress. Considering these factors are closely associated with the pathogenesis of PAH, we hypothesize that TUFM might play a role in the development of PAH. Our preliminary examination has showed TUFM mainly expressed in the PASMCs, and the subsequent test indicated an increased TUFM expression in the SMCs of pulmonary arteriole in monocrotaline- (MCT-) induced PAH rat model compared with the normal rat. The TUFM knockdown (Sh-TUFM) or overexpressed (OE-TUFM) rats were used to establish PAH by treating with MCT. A notable lower pulmonary arterial systolic pressure together with slightly morphological changes of pulmonary arteriole was observed in the Sh-TUFM group compared with the single MCT-induced PAH group. Increased levels of P62 and Bax and reduced LC3II/I, BECN1, and Bcl2 were detected in the Sh-TUFM group, while the expressions of these proteins in the OE-TUFM group were contrast to the results of the Sh-TUFM group. To elucidate the possible mechanism underlying biological effect of TUFM in PAH, PASMCs were treated with silence or overexpression of TUFM and then exposed to hypoxia condition. An obviously high levels of P62 and Bax along with a decreased LC3 II/I, BECN1, ULK1, Atg12, Atg13, and Bcl2 levels were noticed in cells with silence of TUFM. Moreover, the phosphorylated AMPK and mTOR which was well known in mitophagy modulating vary by the alternation of TUFM. These observations suggested that TUFM silence inhibits the development of MCT-induced PAH via AMPK/mTOR pathway.
Copyright © 2022 Ruyuan Wei et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
References
-
- Bernal-Ramírez J., Silva-Platas C., Jerjes-Sánchez C., et al. Resveratrol prevents right ventricle dysfunction, calcium mishandling, and energetic failure via Sirt3 stimulation in pulmonary arterial hypertension. Oxidative Medicine and Cellular Longevity . 2021;2021:15. doi: 10.1155/2021/9912434.9912434 - DOI - PMC - PubMed
-
- Hassoun P. M. Pulmonary arterial hypertension. The New England Journal of Medicine . 2021;385(25):2361–2376. - PubMed
-
- Wanderoy S., Hees J. T., Klesse R., Edlich F., Harbauer A. B. Kill one or kill the many: interplay between mitophagy and apoptosis. Biological Chemistry . 2020;402(1):73–88. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
