

Profiling of epigenetic marker regions in murine ILCs under homeostatic and inflammatory conditions
- PMID: 35938981
- PMCID: PMC9386974
- DOI: 10.1084/jem.20210663
Profiling of epigenetic marker regions in murine ILCs under homeostatic and inflammatory conditions
Abstract
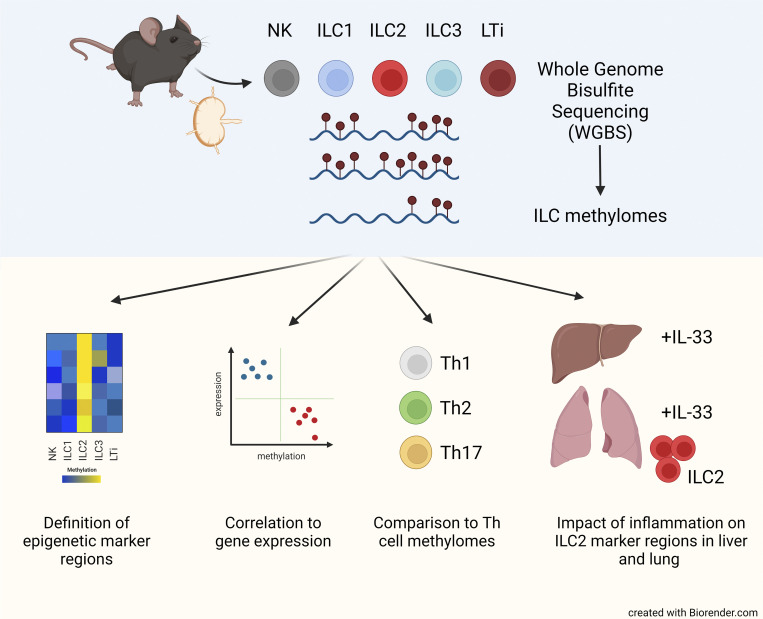
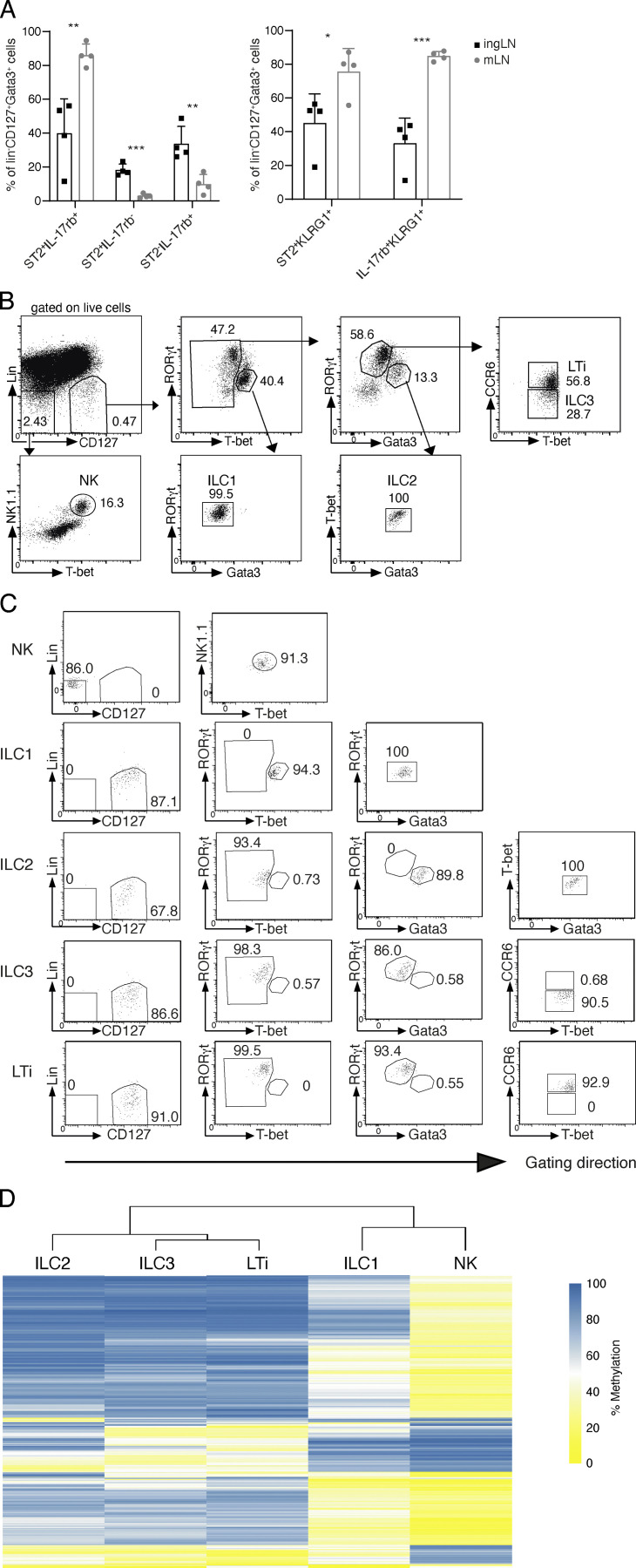
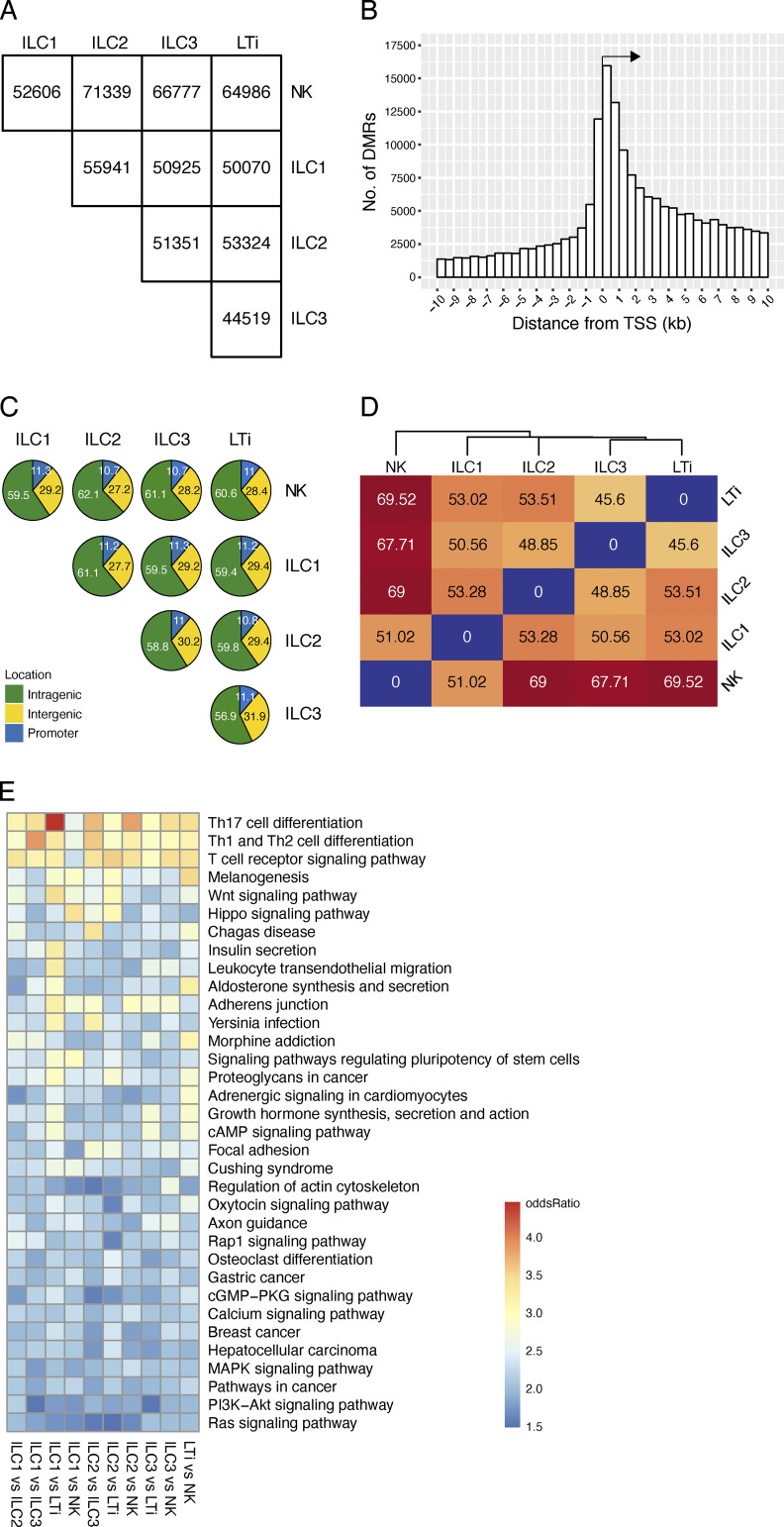
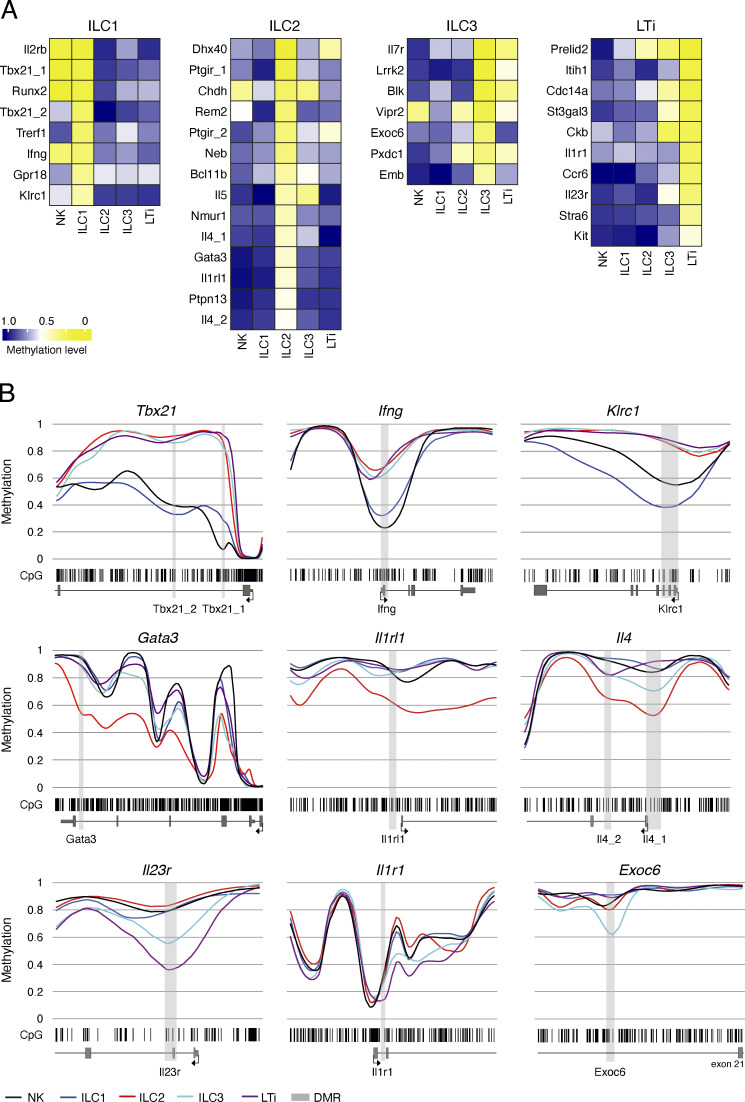
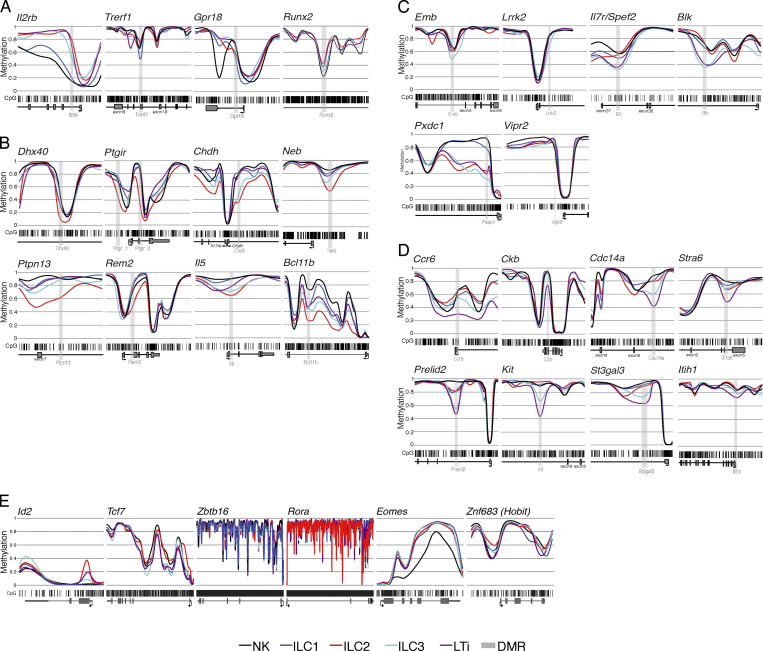
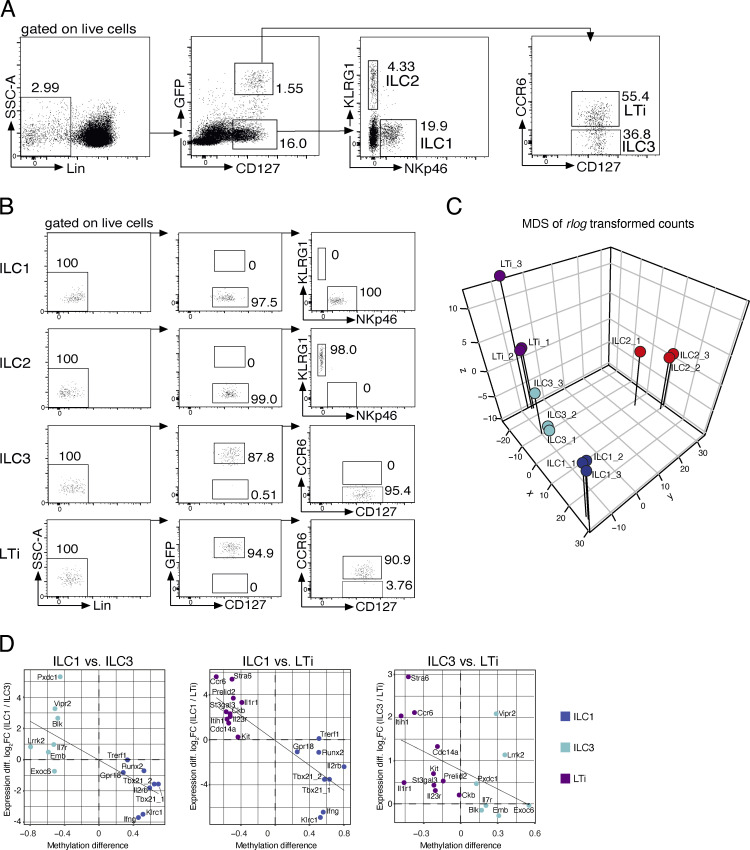
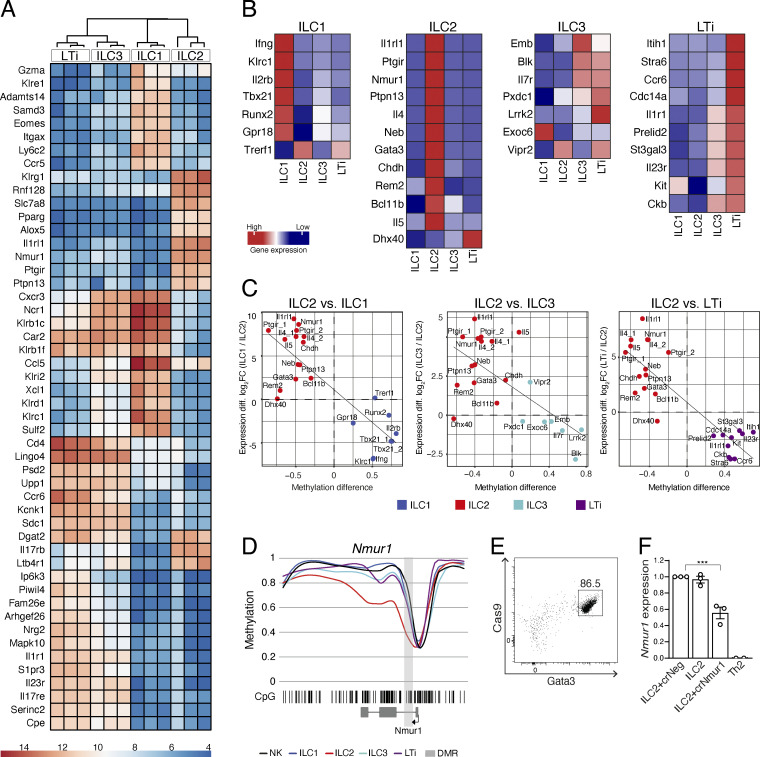
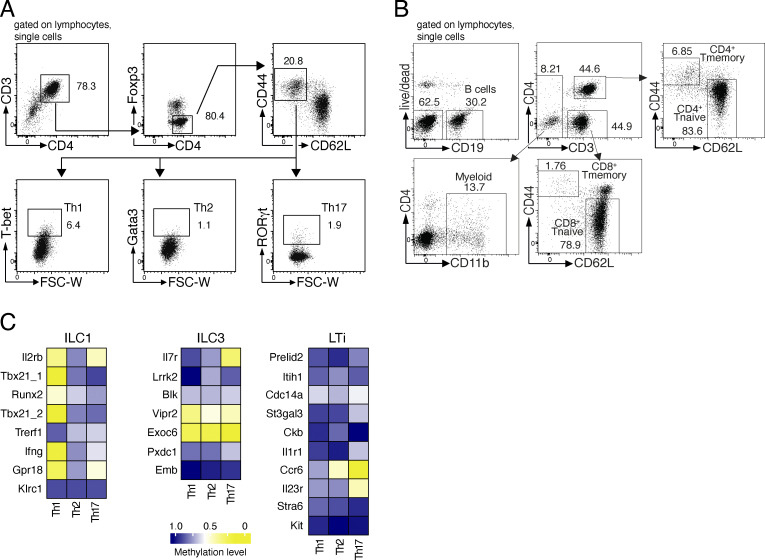
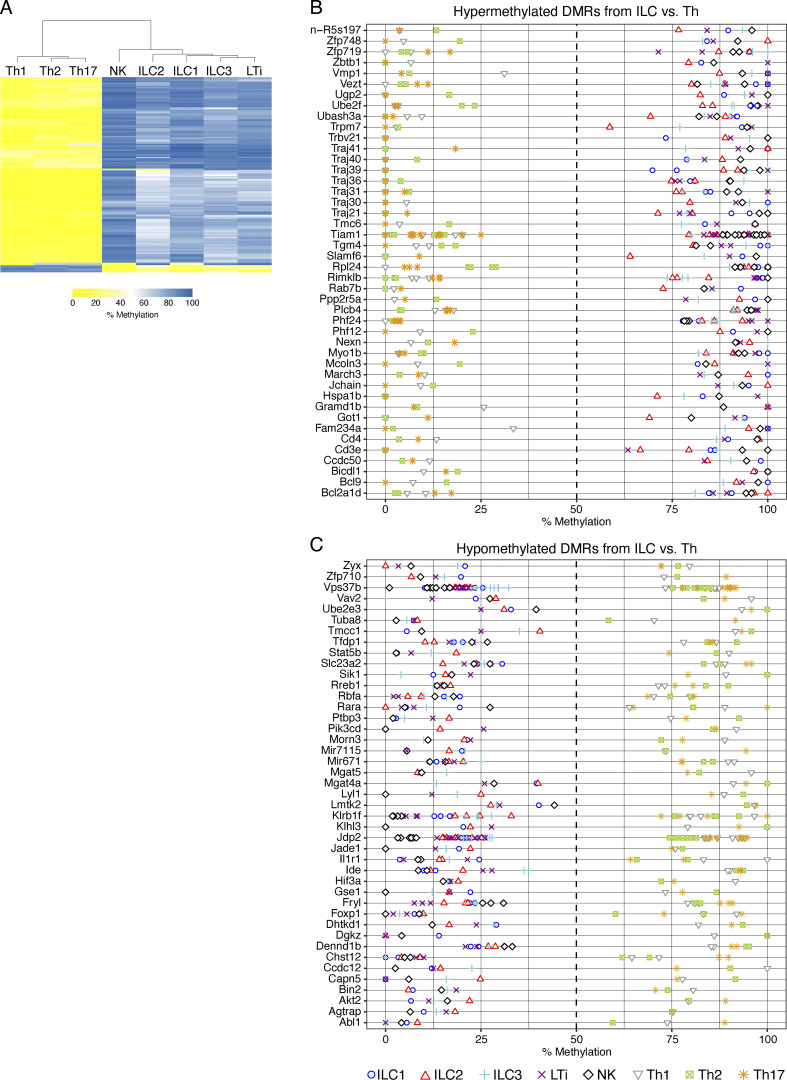
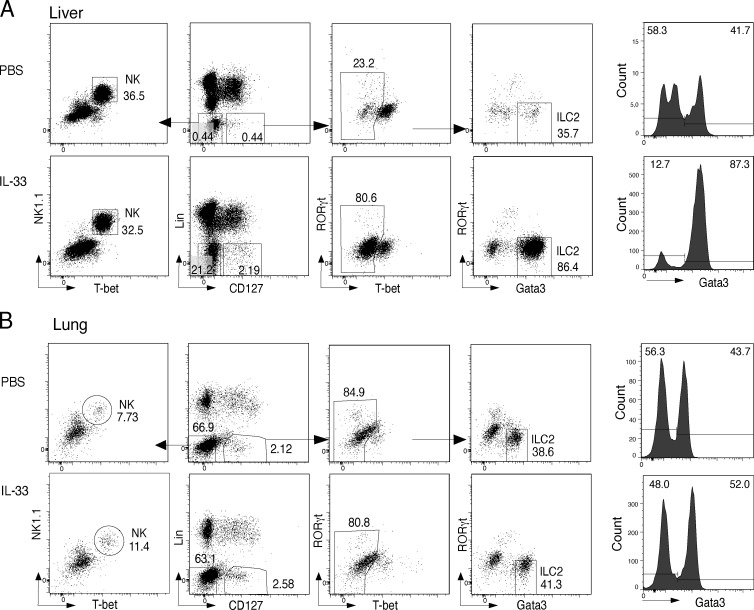
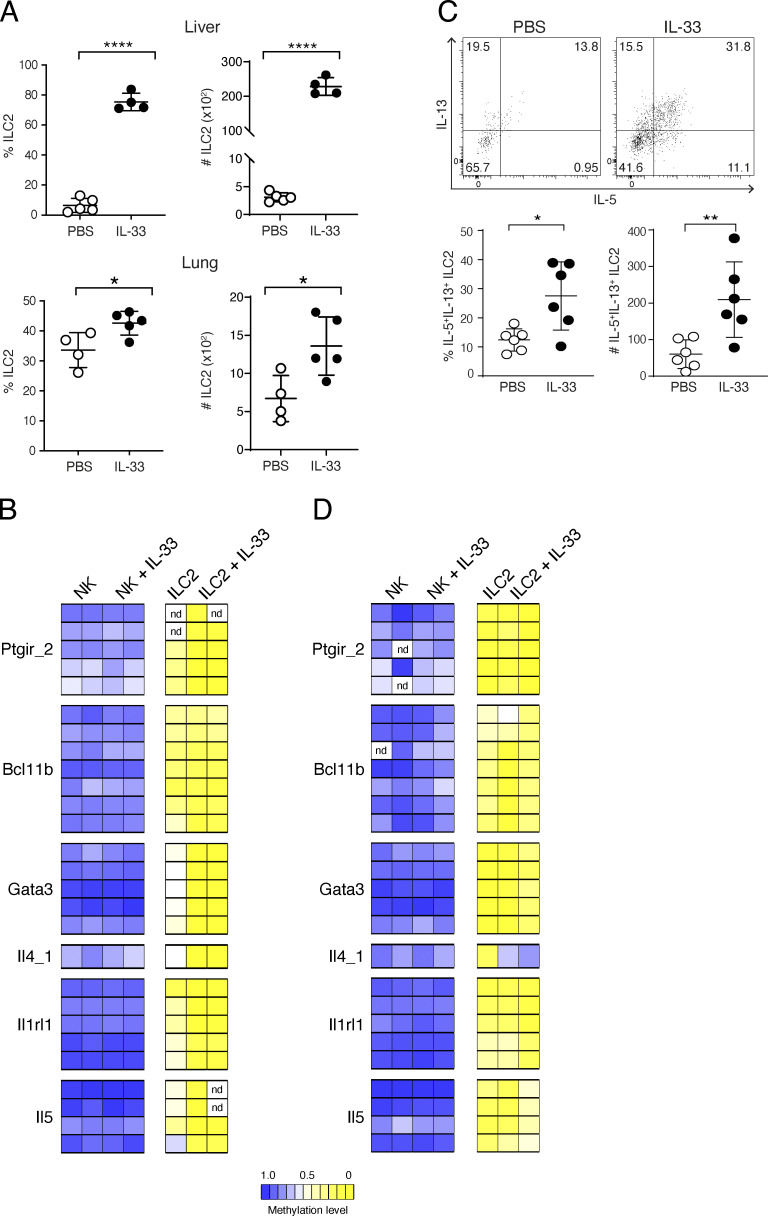
Epigenetic modifications such as DNA methylation play an essential role in imprinting specific transcriptional patterns in cells. We performed genome-wide DNA methylation profiling of murine lymph node-derived ILCs, which led to the identification of differentially methylated regions (DMRs) and the definition of epigenetic marker regions in ILCs. Marker regions were located in genes with a described function for ILCs, such as Tbx21, Gata3, or Il23r, but also in genes that have not been related to ILC biology. Methylation levels of the marker regions and expression of the associated genes were strongly correlated, indicating their functional relevance. Comparison with T helper cell methylomes revealed clear lineage differences, despite partial similarities in the methylation of specific ILC marker regions. IL-33-mediated challenge affected methylation of ILC2 epigenetic marker regions in the liver, while remaining relatively stable in the lung. In our study, we identified a set of epigenetic markers that can serve as a tool to study phenotypic and functional properties of ILCs.
© 2022 Beckstette et al.
Conflict of interest statement
Disclosures: The authors declare no competing interests exist.
Figures

Similar articles
-
Whole-genome profiling of DNA methylation and hydroxymethylation identifies distinct regulatory programs among innate lymphocytes.Nat Immunol. 2022 Apr;23(4):619-631. doi: 10.1038/s41590-022-01164-8. Epub 2022 Mar 24. Nat Immunol. 2022. PMID: 35332328 Free PMC article.
-
The DNA methylation profile of liver tumors in C3H mice and identification of differentially methylated regions involved in the regulation of tumorigenic genes.BMC Cancer. 2018 Mar 22;18(1):317. doi: 10.1186/s12885-018-4221-0. BMC Cancer. 2018. PMID: 29566670 Free PMC article.
-
Epigenetic regulation of innate lymphoid cells.Eur J Immunol. 2024 Sep;54(9):e2350379. doi: 10.1002/eji.202350379. Epub 2024 Jun 2. Eur J Immunol. 2024. PMID: 38824666 Review.
-
Epigenetic profiling at mouse imprinted gene clusters reveals novel epigenetic and genetic features at differentially methylated regions.Genome Res. 2009 Aug;19(8):1374-83. doi: 10.1101/gr.089185.108. Epub 2009 Jun 19. Genome Res. 2009. PMID: 19542493 Free PMC article.
-
Dichotomous Regulation of Acquired Immunity by Innate Lymphoid Cells.Cells. 2020 May 11;9(5):1193. doi: 10.3390/cells9051193. Cells. 2020. PMID: 32403291 Free PMC article. Review.
Cited by
-
cfDNA Methylation Profiles and T-Cell Differentiation in Women with Endometrial Polyps.Cells. 2022 Dec 9;11(24):3989. doi: 10.3390/cells11243989. Cells. 2022. PMID: 36552753 Free PMC article.
-
ILC2-mediated immune crosstalk in chronic (vascular) inflammation.Front Immunol. 2023 Dec 20;14:1326440. doi: 10.3389/fimmu.2023.1326440. eCollection 2023. Front Immunol. 2023. PMID: 38179045 Free PMC article. Review.
-
Differential regulation of lineage-determining transcription factor expression in innate lymphoid cell and adaptive T helper cell subsets.Front Immunol. 2023 Jan 4;13:1081153. doi: 10.3389/fimmu.2022.1081153. eCollection 2022. Front Immunol. 2023. PMID: 36685550 Free PMC article. Review.
-
The IL-23R and Its Genetic Variants: A Hitherto Unforeseen Bridge Between the Immune System and Cancer Development.Cancers (Basel). 2024 Dec 27;17(1):55. doi: 10.3390/cancers17010055. Cancers (Basel). 2024. PMID: 39796684 Free PMC article. Review.
References
-
- Allan, D.S.J., Cerdeira A.S., Ranjan A., Kirkham C.L., Aguilar O.A., Tanaka M., Childs R.W., Dunbar C.E., Strominger J.L., Kopcow H.D., and Carlyle J.R.. 2017. Transcriptome analysis reveals similarities between human blood CD3(−) CD56(bright) cells and mouse CD127(+) innate lymphoid cells. Sci. Rep. 7:3501. 10.1038/s41598-017-03256-0 - DOI - PMC - PubMed
-
- Antignano, F., Braam M., Hughes M.R., Chenery A.L., Burrows K., Gold M.J., Oudhoff M.J., Rattray D., Halim T.Y., Cait A., et al. . 2016. G9a regulates group 2 innate lymphoid cell development by repressing the group 3 innate lymphoid cell program. J. Exp. Med. 213:1153–1162. 10.1084/jem.20151646 - DOI - PMC - PubMed
-
- Bailey, T.L., and Elkan C.. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28–36 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
