

The landscape in the gut microbiome of long-lived families reveals new insights on longevity and aging - relevant neural and immune function
- PMID: 35939616
- PMCID: PMC9361766
- DOI: 10.1080/19490976.2022.2107288
The landscape in the gut microbiome of long-lived families reveals new insights on longevity and aging - relevant neural and immune function
Abstract
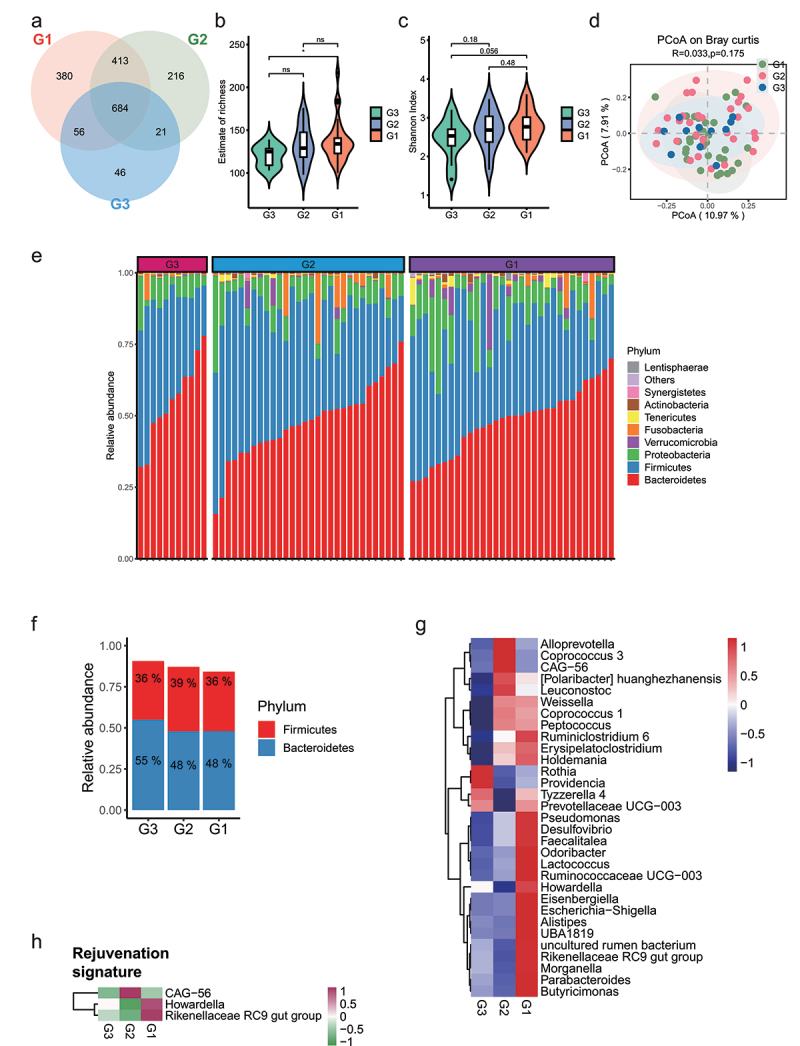
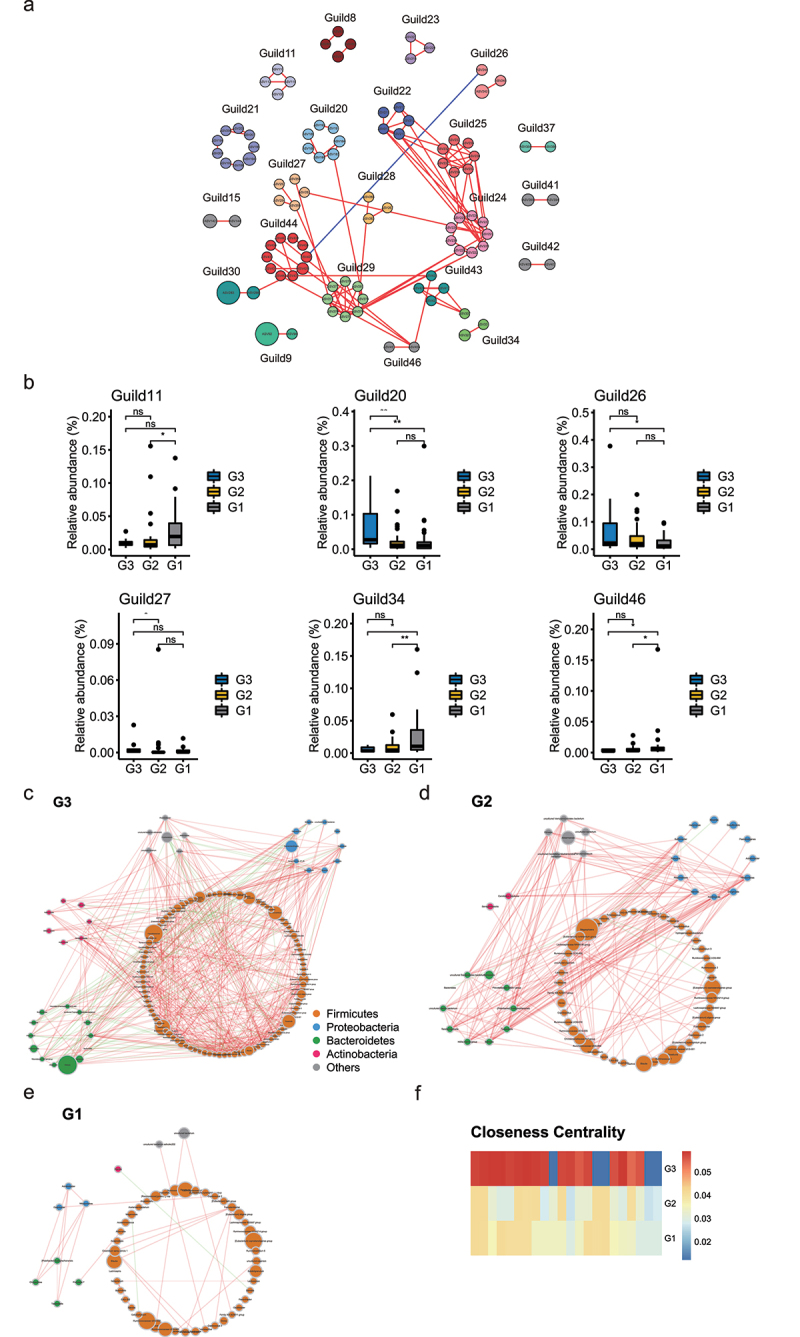
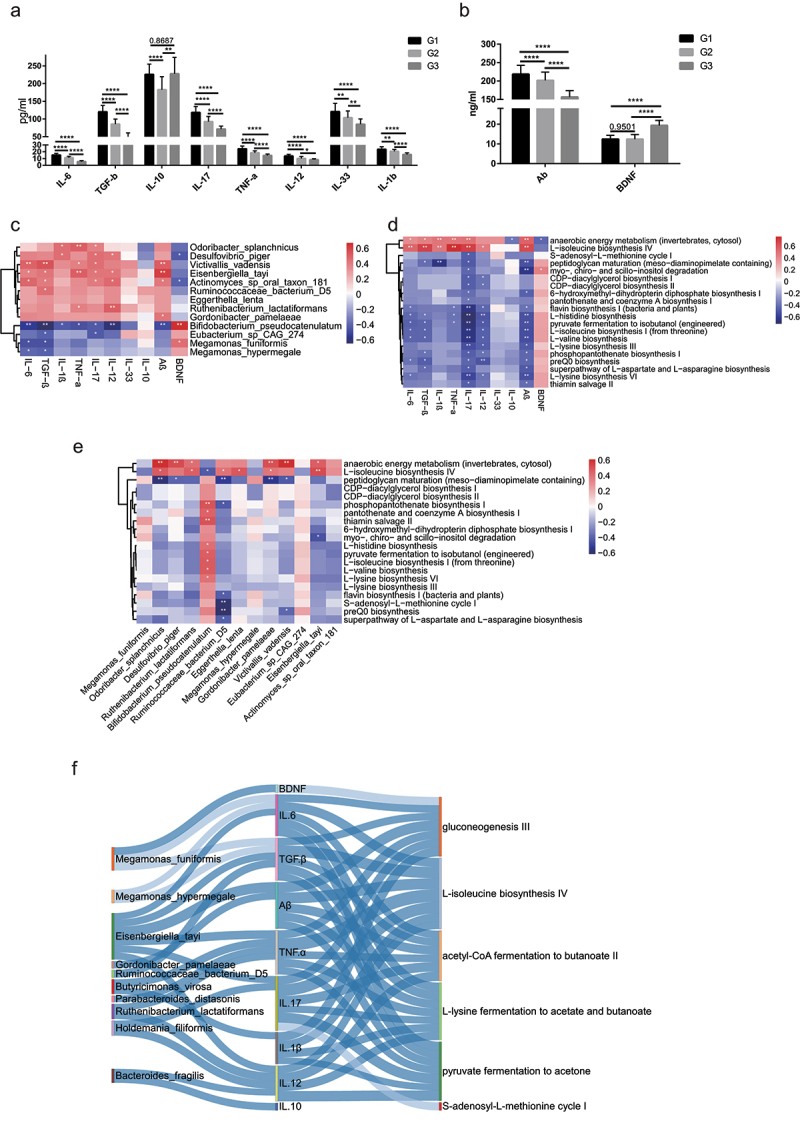
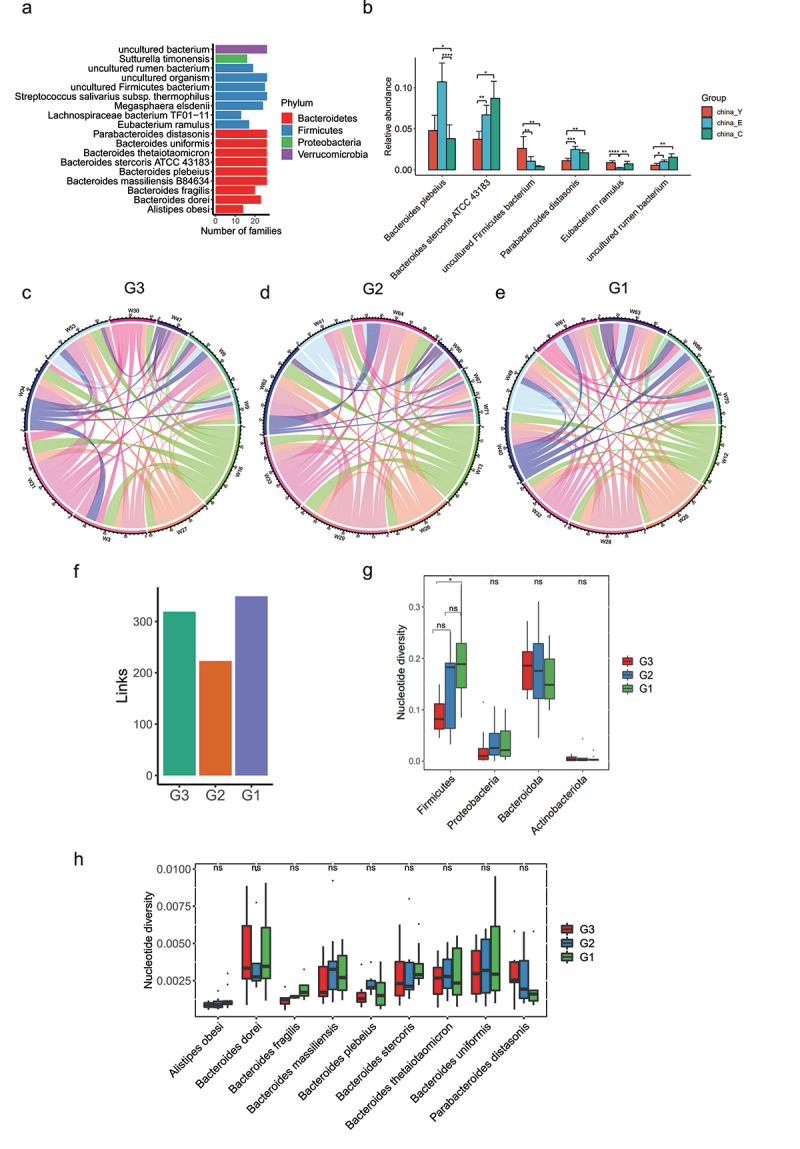
Human longevity has a strong familial and genetic component. Dynamic characteristics of the gut microbiome during aging associated with longevity, neural, and immune function remained unknown. Here, we aim to reveal the synergistic changes in gut microbiome associated with decline in neural and immune system with aging and further obtain insights into the establishment of microbiome homeostasis that can benefit human longevity. Based on 16S rRNA and metagenomics sequencing data for 32 longevity families including three generations, centenarians, elderly, and young groups, we found centenarians showed increased diversity of gut microbiota, severely damaged connection among bacteria, depleted in microbial-associated essential amino acid function, and increased abundance of anti-inflammatory bacteria in comparison to young and elderly groups. Some potential probiotic species, such as Desulfovibrio piger, Gordonibacter pamelaeae, Odoribacter splanchnicus, and Ruminococcaceae bacterium D5 were enriched with aging, which might possibly support health maintenance. The level of Amyloid-β (Aβ) and brain-derived neurotrophic factor (BDNF) related to neural function showed increased and decreased with aging, respectively. The elevated level of inflammatory factors was observed in centenarians compared with young and elderly groups. The enriched Bacteroides fragilis in centenarians might promote longevity through up-regulating anti-inflammatory factor IL-10 expression to mediate the critical balance between health and disease. Impressively, the associated analysis for gut microbiota with the level of Aβ, BDNF, and inflammatory factors suggests Bifidobacterium pseudocatenulatum could be a particularly beneficial bacteria in the improvement of impaired neural and immune function. Our results provide a rationale for targeting the gut microbiome in future clinical applications of aging-related diseases and extending life span.Abbreviations: 16S rRNA: 16S ribosomal RNA; MAGs: Metagenome-assembled genomes; ASVs: Amplicon sequence variants; DNA: Deoxyribonucleic acid; FDR: False discovery rate: KEGG: Kyoto Encyclopedia of Genes and Genomes; PCoA: Principal coordinates analysis; PCR: Polymerase chain reaction; PICRUSt: Phylogenetic Investigation of Communities by Reconstruction of Unobserved States; Aβ: Amyloid-β (Aβ); BDNF: Brain-derived neurotrophic factor.
Keywords: Centenarians; bifidobacterium pseudocatenulatum; gut microbiome; longevity; odoribacter splanchnicus.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases