

cfDNA methylome profiling for detection and subtyping of small cell lung cancers
- PMID: 35941262
- PMCID: PMC9586870
- DOI: 10.1038/s43018-022-00415-9
cfDNA methylome profiling for detection and subtyping of small cell lung cancers
Abstract
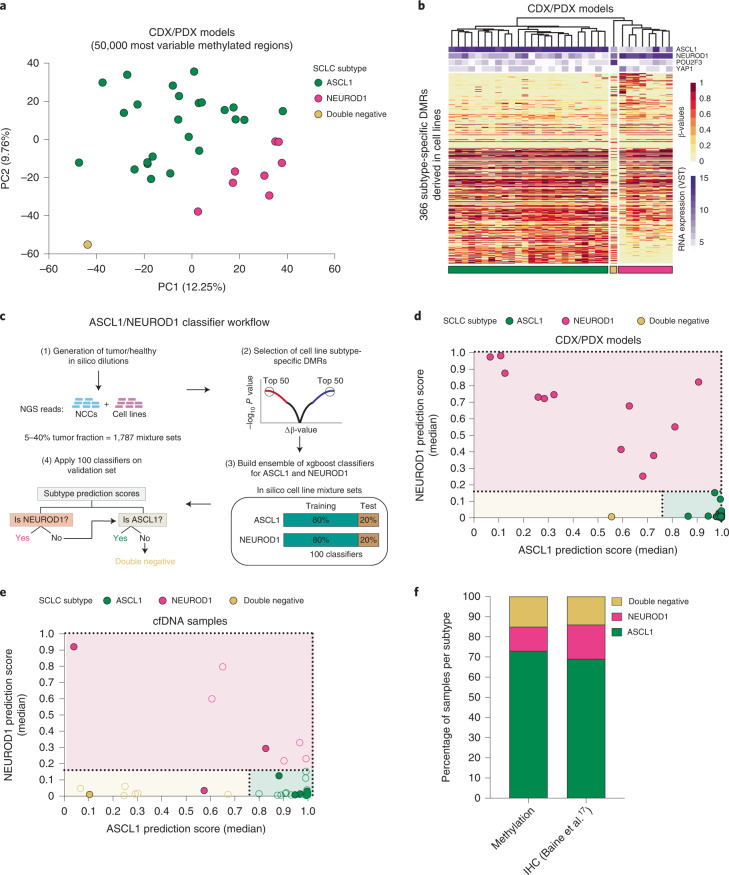
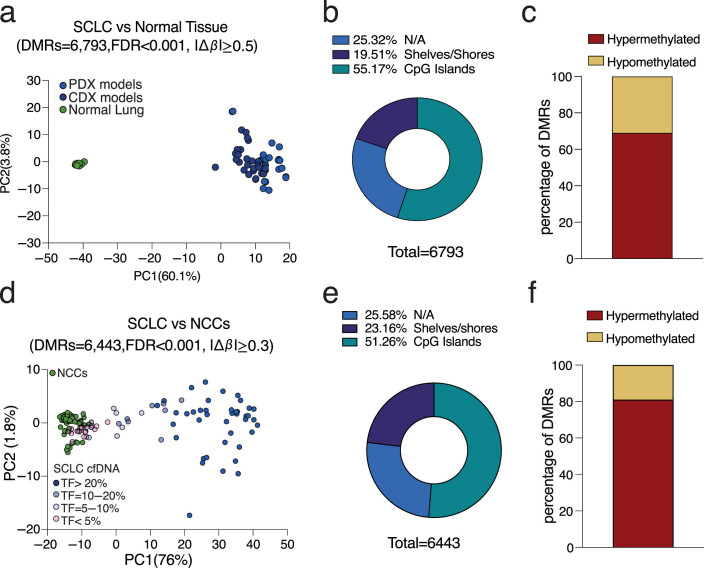
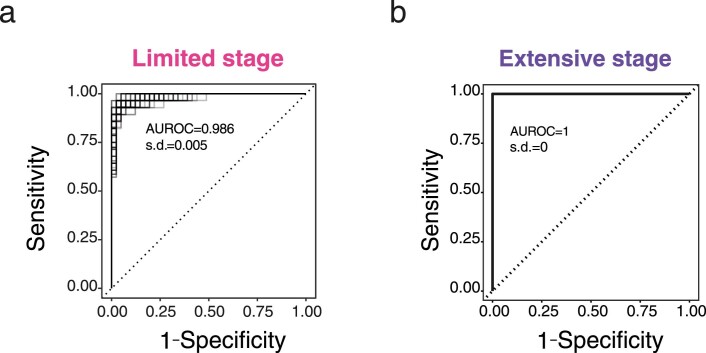
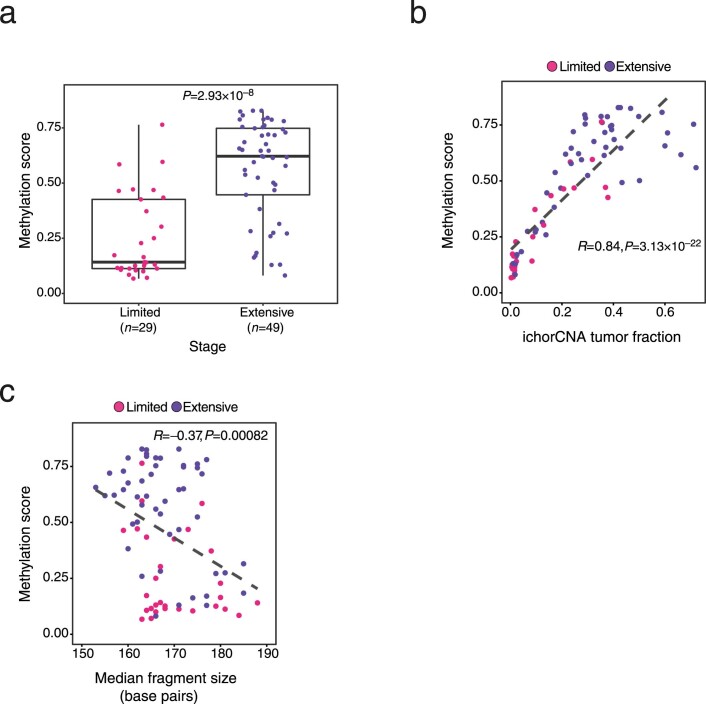
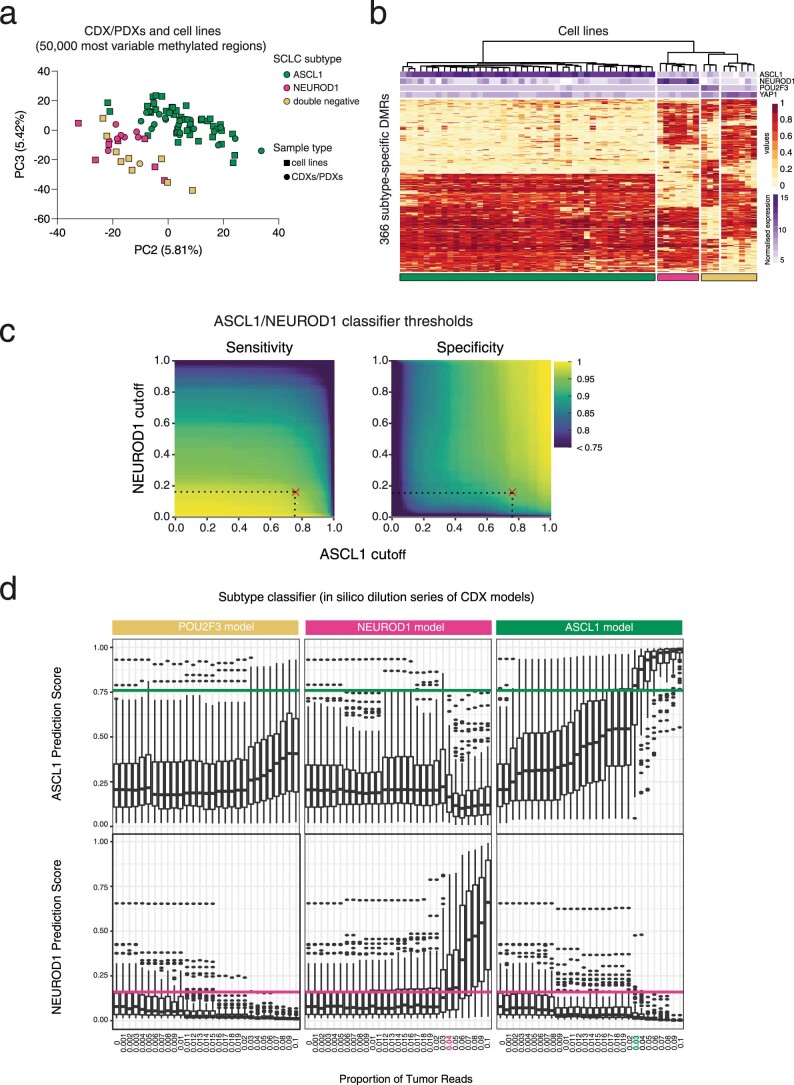
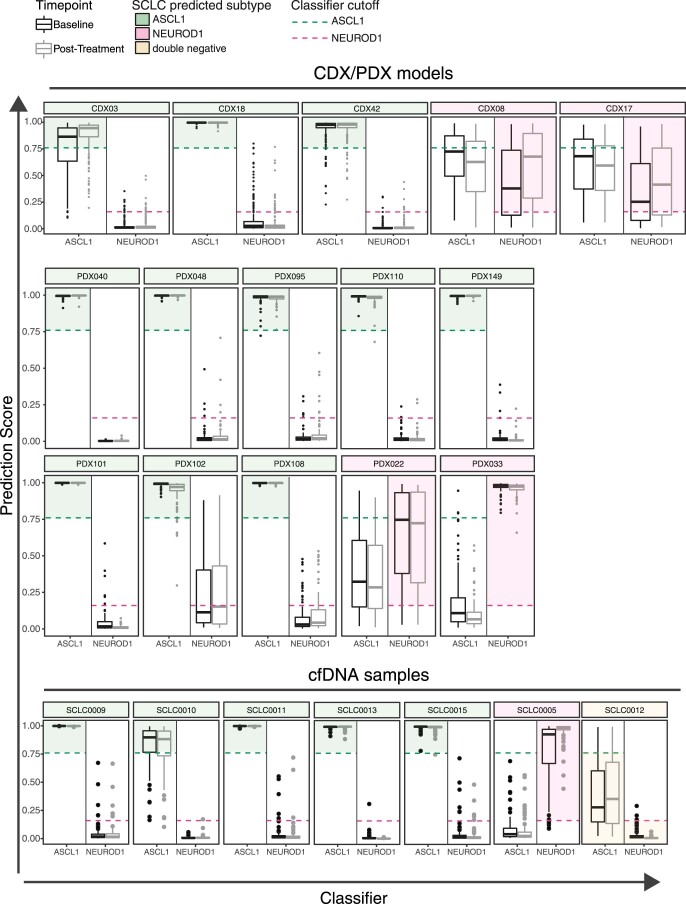
Small cell lung cancer (SCLC) is characterized by morphologic, epigenetic and transcriptomic heterogeneity. Subtypes based upon predominant transcription factor expression have been defined that, in mouse models and cell lines, exhibit potential differential therapeutic vulnerabilities, with epigenetically distinct SCLC subtypes also described. The clinical relevance of these subtypes is unclear, due in part to challenges in obtaining tumor biopsies for reliable profiling. Here we describe a robust workflow for genome-wide DNA methylation profiling applied to both patient-derived models and to patients' circulating cell-free DNA (cfDNA). Tumor-specific methylation patterns were readily detected in cfDNA samples from patients with SCLC and were correlated with survival outcomes. cfDNA methylation also discriminated between the transcription factor SCLC subtypes, a precedent for a liquid biopsy cfDNA-methylation approach to molecularly subtype SCLC. Our data reveal the potential clinical utility of cfDNA methylation profiling as a universally applicable liquid biopsy approach for the sensitive detection, monitoring and molecular subtyping of patients with SCLC.
© 2022. The Author(s).
Conflict of interest statement
C.M.R. has consulted regarding oncology drug development with AbbVie, Amgen, Astra Zeneca, Epizyme, Genentech/Roche, Ipsen, Jazz, Lilly and Syros and serves on the scientific advisory boards of Bridge Medicines, Earli and Harpoon Therapeutics. C.D. receives research grants/support from AstraZeneca, Astex Pharmaceuticals, Bioven, Amgen, Carrick Therapeutics, Merck AG, Taiho Oncology, GSK, Bayer, Boehringer Ingelheim, Roche, BMS, Novartis, Celgene, Epigene Therapeutics, Angle, Menarini, Clearbridge Biomedics, Thermo Fisher Scientific and Neomed Therapeutics. C.D. has received honoraria/consultancy fees from Biocartis, Merck, AstraZeneca and GRAIL. The remaining authors declare no competing interests.
Figures
References
-
- Horn L, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N. Engl. J. Med. 2018;379:2220–2229. - PubMed
-
- Gazdar AF, Carney DN, Nau MM, Minna JD. Characterization of variant subclasses of cell lines derived from small cell lung cancer having distinctive biochemical, morphological, and growth properties. Cancer Res. 1985;45:2924–2930. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
