

Patterns of structural variation define prostate cancer across disease states
- PMID: 35943799
- PMCID: PMC9536266
- DOI: 10.1172/jci.insight.161370
Patterns of structural variation define prostate cancer across disease states
Abstract
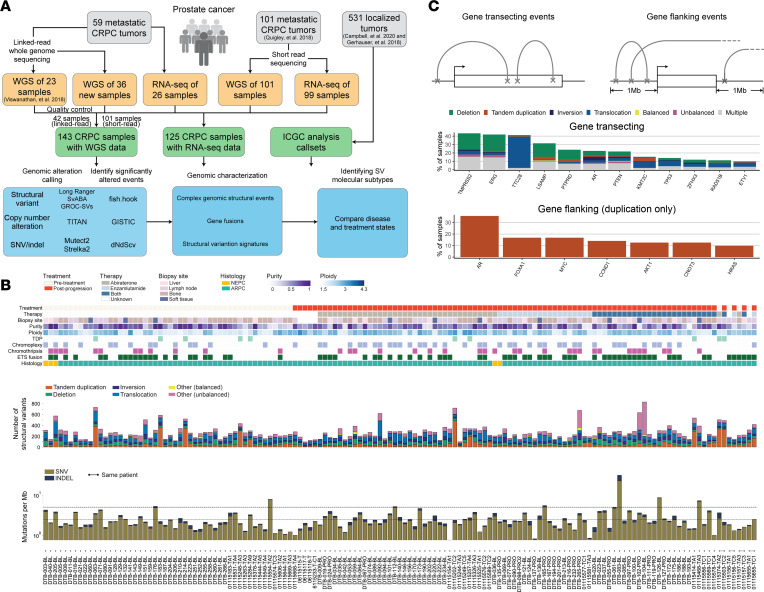
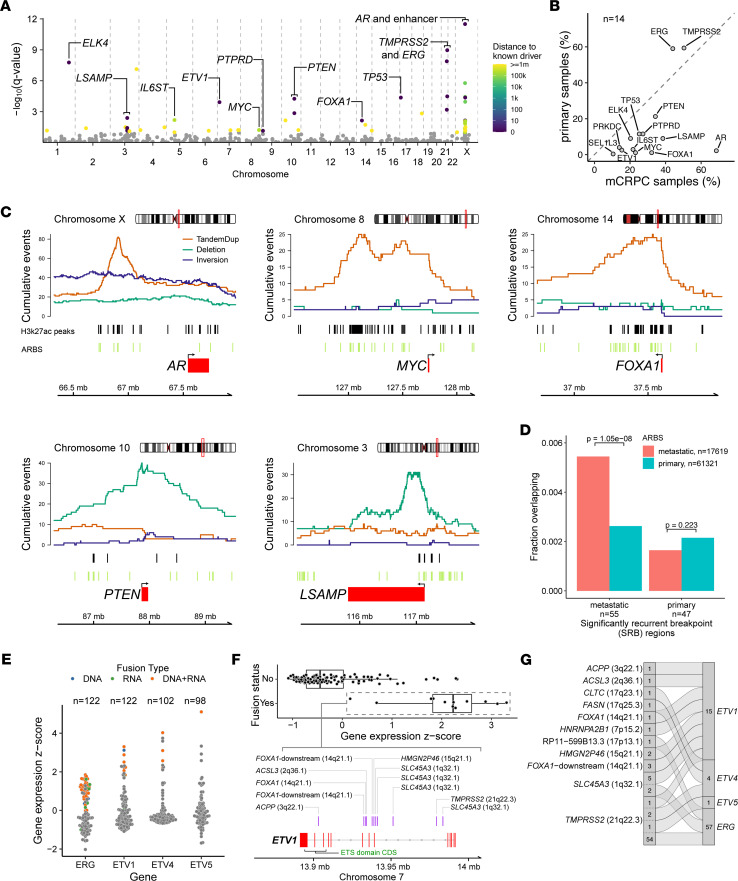
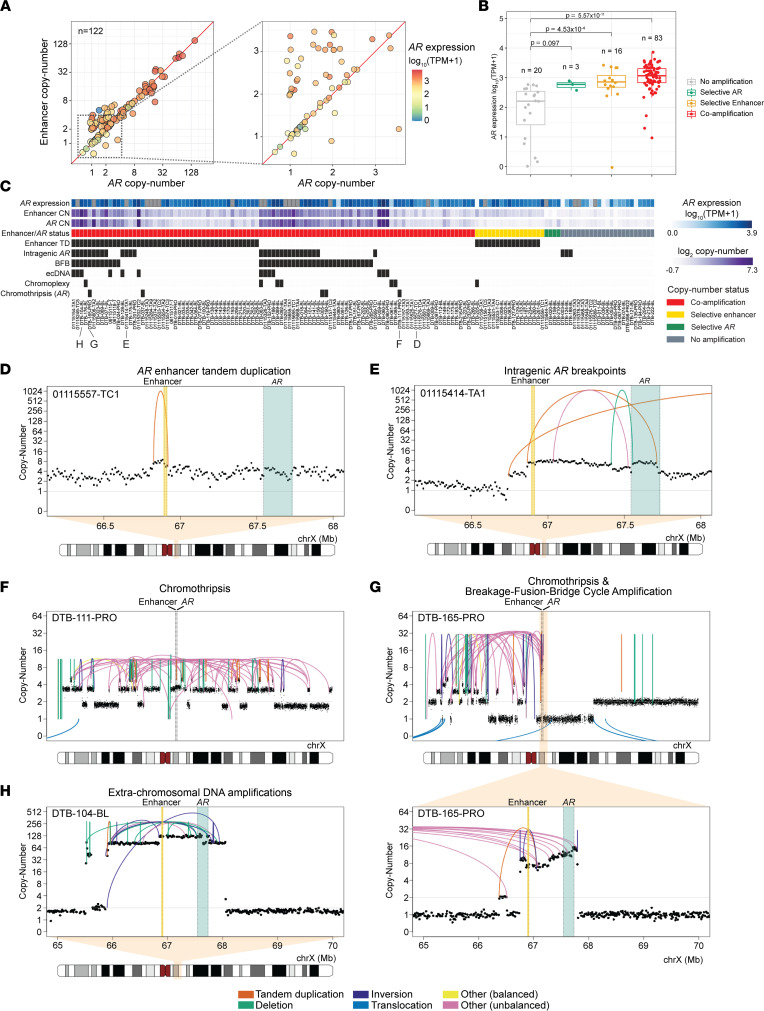
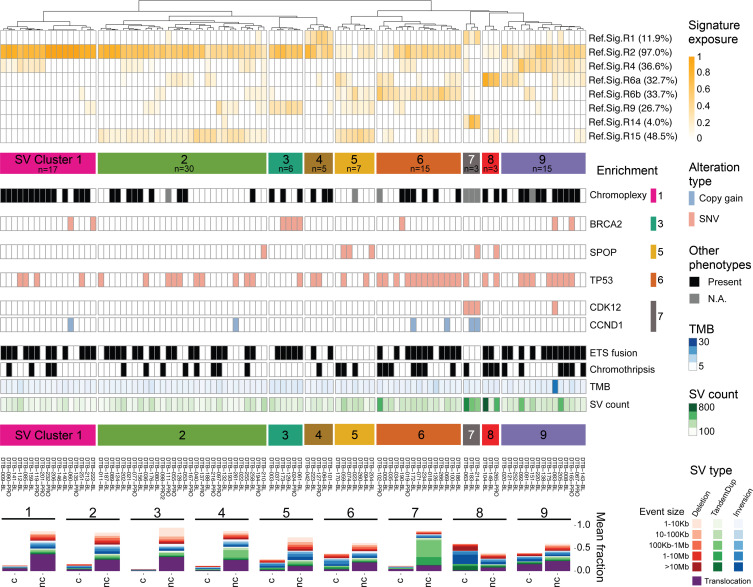
The complex genomic landscape of prostate cancer evolves across disease states under therapeutic pressure directed toward inhibiting androgen receptor (AR) signaling. While significantly altered genes in prostate cancer have been extensively defined, there have been fewer systematic analyses of how structural variation shapes the genomic landscape of this disease across disease states. We uniformly characterized structural alterations across 531 localized and 143 metastatic prostate cancers profiled by whole genome sequencing, 125 metastatic samples of which were also profiled via whole transcriptome sequencing. We observed distinct significantly recurrent breakpoints in localized and metastatic castration-resistant prostate cancers (mCRPC), with pervasive alterations in noncoding regions flanking the AR, MYC, FOXA1, and LSAMP genes enriched in mCRPC and TMPRSS2-ERG rearrangements enriched in localized prostate cancer. We defined 9 subclasses of mCRPC based on signatures of structural variation, each associated with distinct genetic features and clinical outcomes. Our results comprehensively define patterns of structural variation in prostate cancer and identify clinically actionable subgroups based on whole genome profiling.
Keywords: Bioinformatics; Genetic variation; Genetics; Oncology; Prostate cancer.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
