

PD 102807 Induces M3 mAChR-Dependent GRK-/Arrestin-Biased Signaling in Airway Smooth Muscle Cells
- PMID: 35944139
- PMCID: PMC9651198
- DOI: 10.1165/rcmb.2021-0320OC
PD 102807 Induces M3 mAChR-Dependent GRK-/Arrestin-Biased Signaling in Airway Smooth Muscle Cells
Abstract
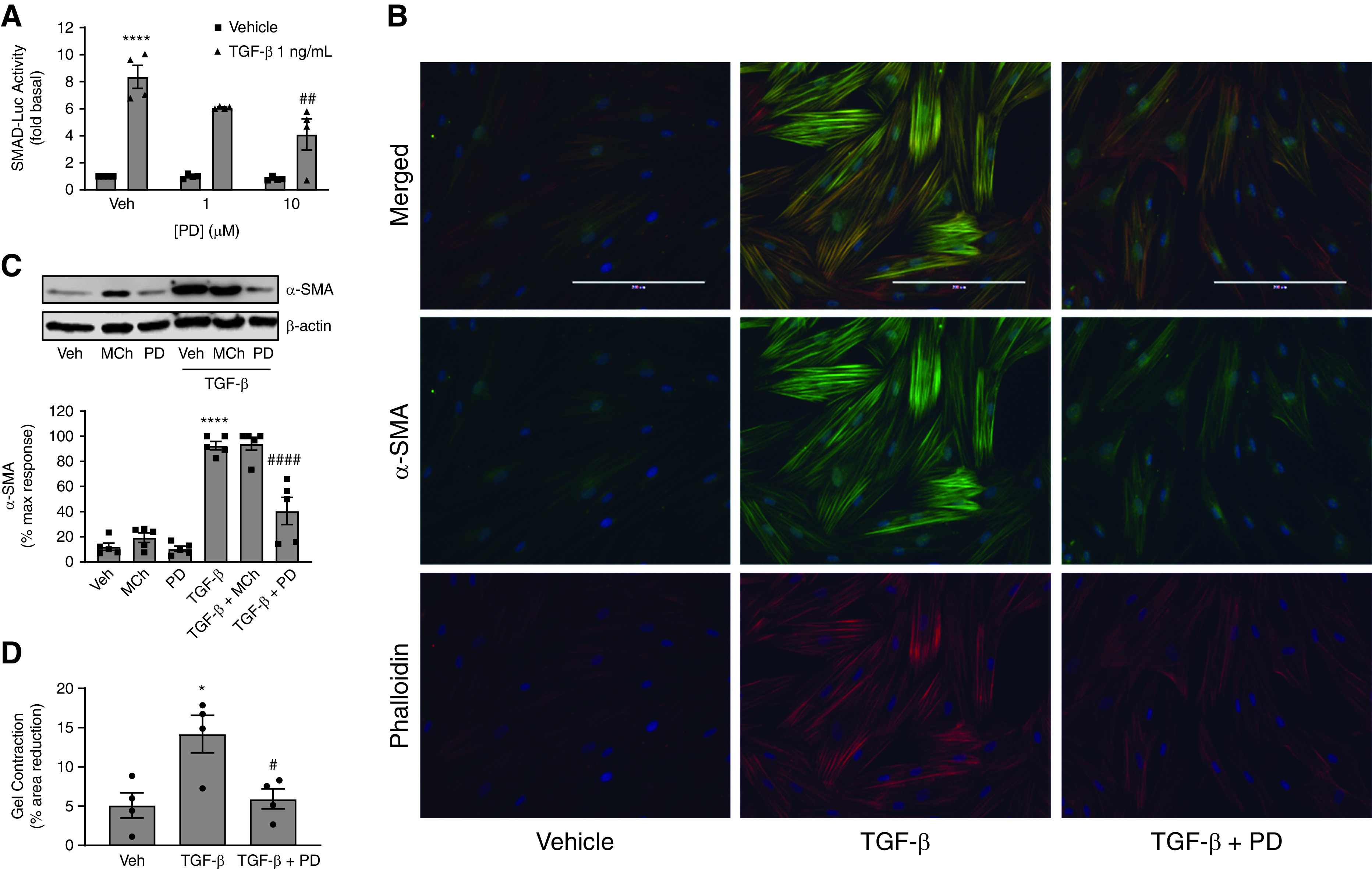
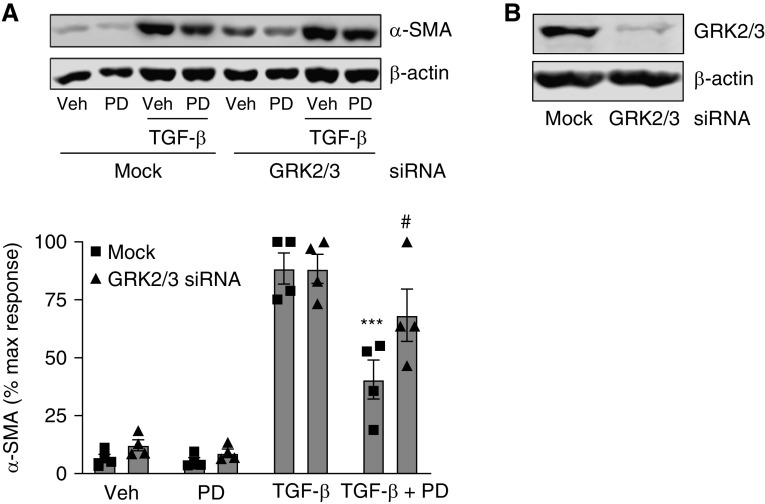
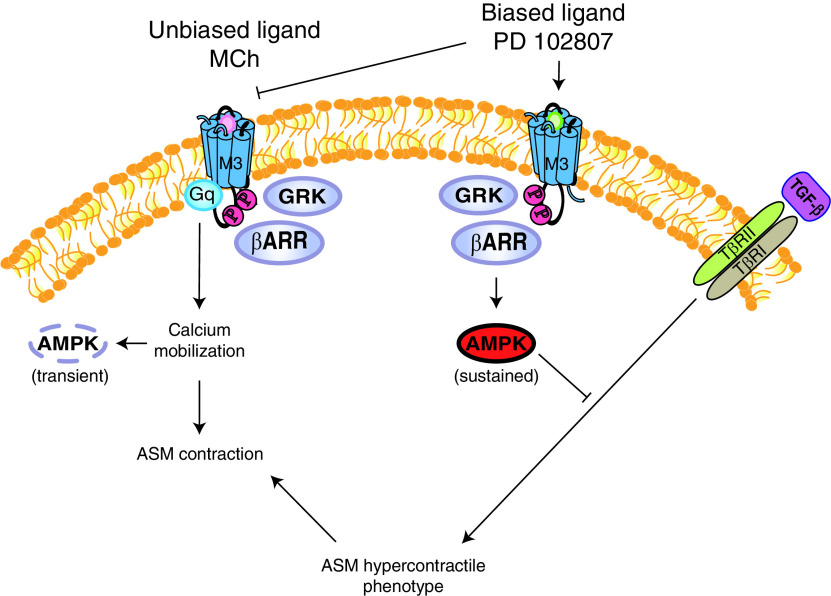
G protein-coupled receptors (GPCRs) not only are turned on or off to control canonical G protein signaling but also may be fine-tuned to promote qualitative/biased signaling. Qualitative signaling by M3 muscarinic acetylcholine receptors (mAChRs) has been proposed, but its impact on physiologic systems remains unclear, and currently no biased M3 mAChR ligands have been described. Herein, we identify PD 102807 as a biased M3 ligand and delineate its signaling and function in human airway smooth muscle (ASM) cells. PD 102807 induced M3-mediated β-arrestin recruitment but not calcium mobilization. PD 102807 inhibited methacholine (MCh)-induced calcium mobilization in (M3-expressing) ASM cells. PD 102807 induced phosphorylation of AMP-activated protein kinase (AMPK) and the downstream effector acetyl-coenzyme A carboxylase (ACC). PD 102807- induced phosphorylated (p)-AMPK levels were greatly reduced in ASM cells with minimal M3 expression and were not inhibited by the Gq inhibitor YM-254890. Induction of p-AMPK and p-ACC was inhibited by β-arrestin 1 or GRK2/3 knockdown. Similarly, MCh induced phosphorylation of AMPK/ACC, but these effects were Gq dependent and unaffected by GRK2/3 knockdown. Consistent with the known ability of AMPK to inhibit transforming growth factor β (TGF-β)-mediated functions, PD 102807 inhibited TGF-β-induced SMAD-Luc activity, sm-α-actin expression, actin stress fiber formation, and ASM cell hypercontractility. These findings reveal that PD 102807 is a biased M3 ligand that inhibits M3-transduced Gq signaling but promotes Gq protein-independent, GRK-/arrestin-dependent, M3-mediated AMPK signaling, which in turn regulates ASM phenotype and contractile function. Consequently, biased M3 ligands hold significant promise as therapeutic agents capable of exploiting the pleiotropic nature of M3 signaling.
Keywords: COPD; acetylcholine; asthma; biased signaling.
Figures
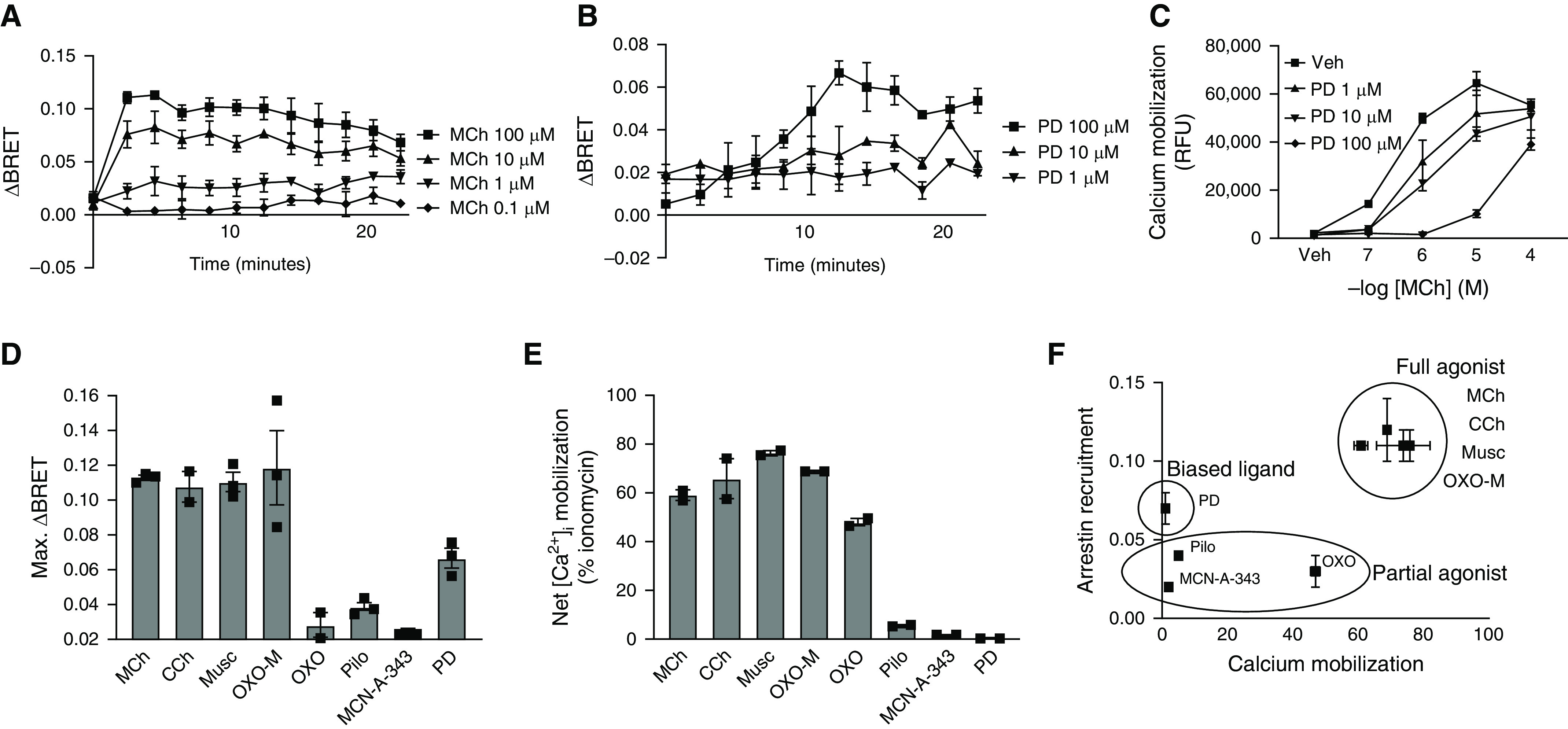
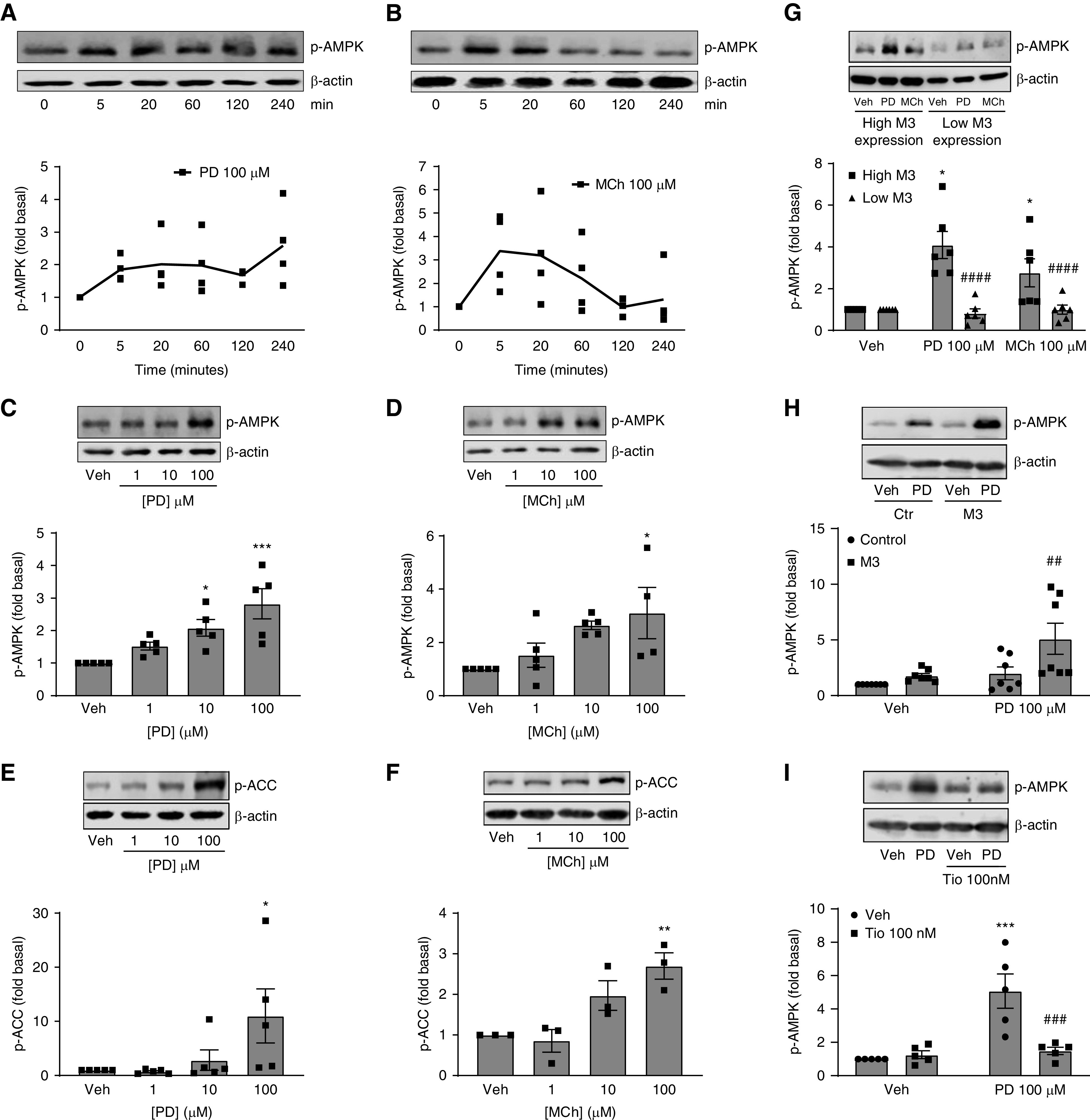
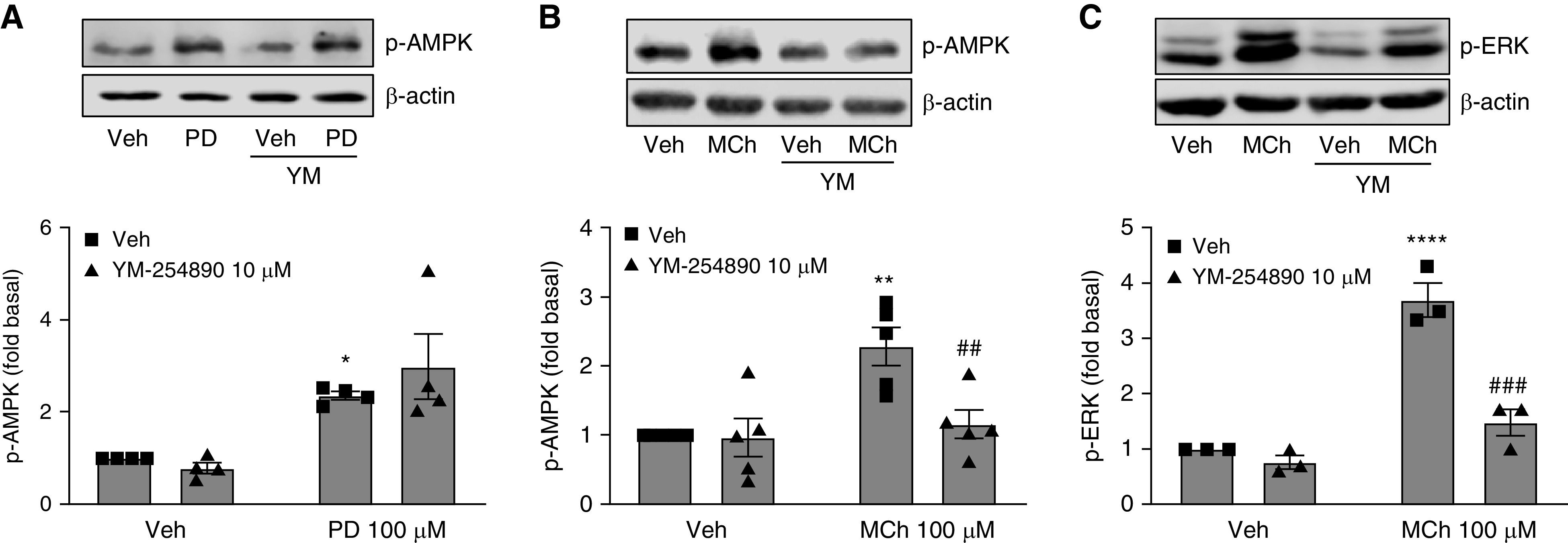
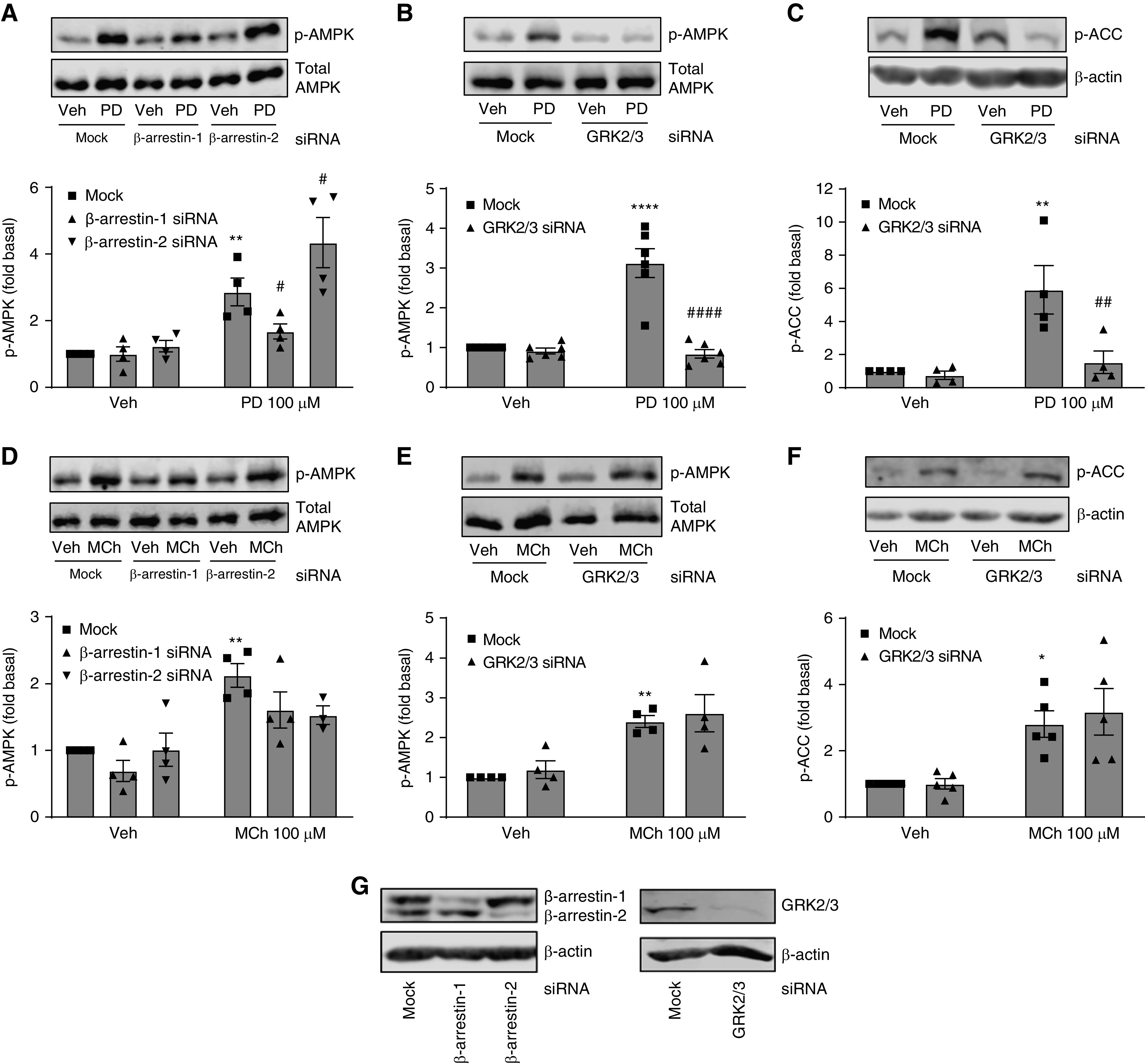
Comment in
-
New Tricks for an Old Dog: Biased GPCR Agonism of an M4 Muscarinic Acetylcholine Receptor Antagonist in Airway Smooth Muscle Cells.Am J Respir Cell Mol Biol. 2022 Nov;67(5):515-517. doi: 10.1165/rcmb.2022-0335ED. Am J Respir Cell Mol Biol. 2022. PMID: 36049223 Free PMC article. No abstract available.
Similar articles
-
The biased M3 mAChR ligand PD 102807 mediates qualitatively distinct signaling to regulate airway smooth muscle phenotype.J Biol Chem. 2023 Oct;299(10):105209. doi: 10.1016/j.jbc.2023.105209. Epub 2023 Sep 1. J Biol Chem. 2023. PMID: 37660916 Free PMC article.
-
Specificity of arrestin subtypes in regulating airway smooth muscle G protein-coupled receptor signaling and function.FASEB J. 2015 Oct;29(10):4227-35. doi: 10.1096/fj.15-273094. Epub 2015 Jun 23. FASEB J. 2015. PMID: 26103985 Free PMC article.
-
Muscarinic receptor stimulation augments TGF-β1-induced contractile protein expression by airway smooth muscle cells.Am J Physiol Lung Cell Mol Physiol. 2012 Oct 1;303(7):L589-97. doi: 10.1152/ajplung.00400.2011. Epub 2012 Aug 3. Am J Physiol Lung Cell Mol Physiol. 2012. PMID: 22865549
-
Novel non-canonical TGF-β signaling networks: emerging roles in airway smooth muscle phenotype and function.Pulm Pharmacol Ther. 2013 Feb;26(1):50-63. doi: 10.1016/j.pupt.2012.07.006. Epub 2012 Jul 31. Pulm Pharmacol Ther. 2013. PMID: 22874922 Review.
-
Biosensor Assays for Measuring the Kinetics of G-Protein and Arrestin-Mediated Signaling in Live Cells.2021 Sep 1. In: Markossian S, Grossman A, Baskir H, Arkin M, Auld D, Austin C, Baell J, Brimacombe K, Chung TDY, Coussens NP, Dahlin JL, Devanarayan V, Foley TL, Glicksman M, Gorshkov K, Grotegut S, Hall MD, Hoare S, Inglese J, Iversen PW, Lal-Nag M, Li Z, Manro JR, McGee J, Norvil A, Pearson M, Riss T, Saradjian P, Sittampalam GS, Tarselli MA, Trask OJ Jr, Weidner JR, Wildey MJ, Wilson K, Xia M, Xu X, editors. Assay Guidance Manual [Internet]. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004–. 2021 Sep 1. In: Markossian S, Grossman A, Baskir H, Arkin M, Auld D, Austin C, Baell J, Brimacombe K, Chung TDY, Coussens NP, Dahlin JL, Devanarayan V, Foley TL, Glicksman M, Gorshkov K, Grotegut S, Hall MD, Hoare S, Inglese J, Iversen PW, Lal-Nag M, Li Z, Manro JR, McGee J, Norvil A, Pearson M, Riss T, Saradjian P, Sittampalam GS, Tarselli MA, Trask OJ Jr, Weidner JR, Wildey MJ, Wilson K, Xia M, Xu X, editors. Assay Guidance Manual [Internet]. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004–. PMID: 34606191 Free Books & Documents. Review.
Cited by
-
Integrative Roles of Pro-Inflammatory Cytokines on Airway Smooth Muscle Structure and Function in Asthma.Immunol Rev. 2025 Mar;330(1):e70007. doi: 10.1111/imr.70007. Immunol Rev. 2025. PMID: 39991781 Free PMC article. Review.
-
Diacylglycerol kinase is a keystone regulator of signaling relevant to the pathophysiology of asthma.Am J Physiol Lung Cell Mol Physiol. 2024 Jul 1;327(1):L3-L18. doi: 10.1152/ajplung.00091.2024. Epub 2024 May 14. Am J Physiol Lung Cell Mol Physiol. 2024. PMID: 38742284 Free PMC article. Review.
-
N-cadherin antagonism is bronchoprotective in severe asthma models.Sci Adv. 2024 Nov 29;10(48):eadp8872. doi: 10.1126/sciadv.adp8872. Epub 2024 Nov 29. Sci Adv. 2024. PMID: 39612338 Free PMC article.
-
Interleukin 31 receptor α promotes smooth muscle cell contraction and airway hyperresponsiveness in asthma.Nat Commun. 2023 Dec 11;14(1):8207. doi: 10.1038/s41467-023-44040-1. Nat Commun. 2023. PMID: 38081868 Free PMC article.
-
New Tricks for an Old Dog: Biased GPCR Agonism of an M4 Muscarinic Acetylcholine Receptor Antagonist in Airway Smooth Muscle Cells.Am J Respir Cell Mol Biol. 2022 Nov;67(5):515-517. doi: 10.1165/rcmb.2022-0335ED. Am J Respir Cell Mol Biol. 2022. PMID: 36049223 Free PMC article. No abstract available.
References
-
- McDonald PH, Lefkowitz RJ. Beta-arrestins: new roles in regulating heptahelical receptors’ functions. Cell Signal . 2001;13:683–689. - PubMed
-
- Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. Beta-arrestin-biased agonism at the beta2-adrenergic receptor. J Biol Chem . 2008;283:5669–5676. - PubMed
-
- Benovic JL, Kühn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein) Proc Natl Acad Sci USA . 1987;84:8879–8882. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
