

Rapid and In-Depth Coverage of the (Phospho-)Proteome With Deep Libraries and Optimal Window Design for dia-PASEF
- PMID: 35944843
- PMCID: PMC9465115
- DOI: 10.1016/j.mcpro.2022.100279
Rapid and In-Depth Coverage of the (Phospho-)Proteome With Deep Libraries and Optimal Window Design for dia-PASEF
Abstract
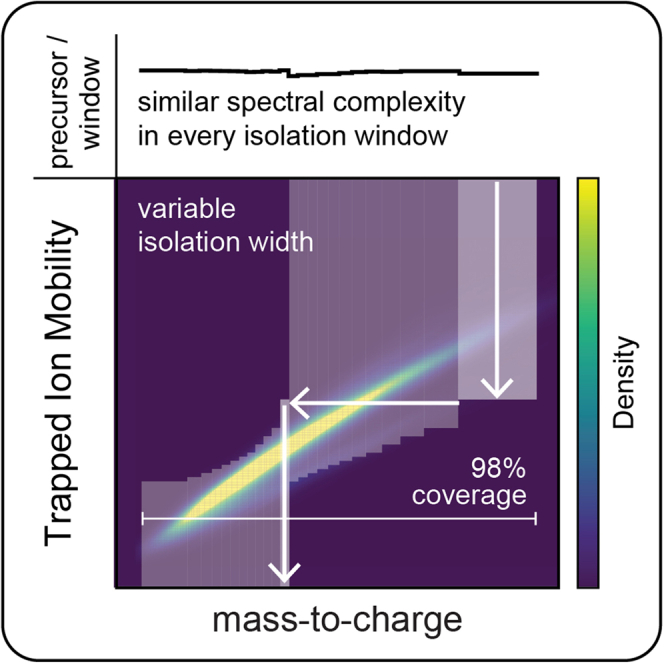
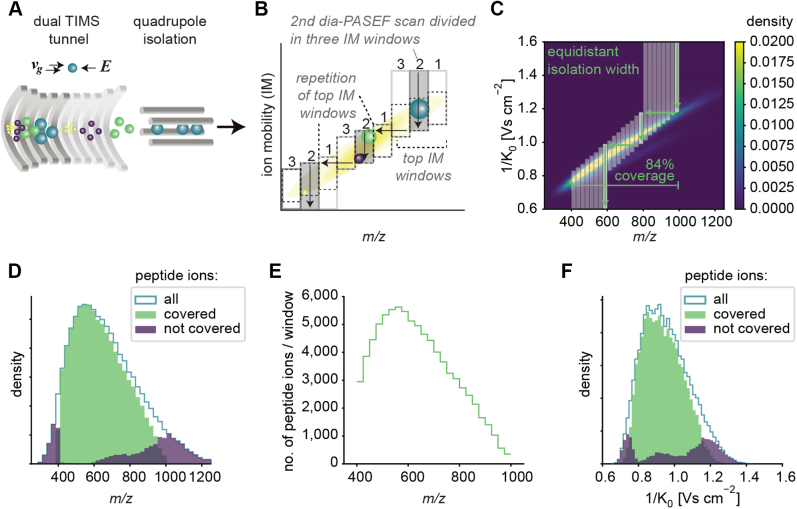
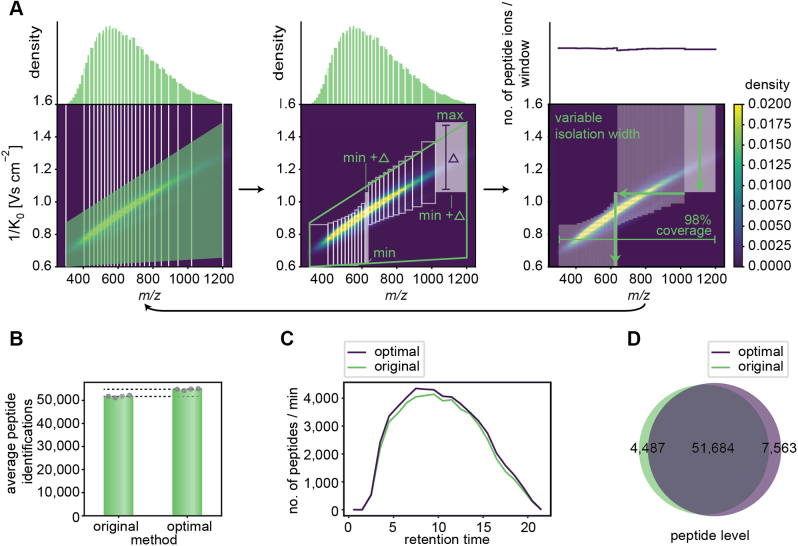
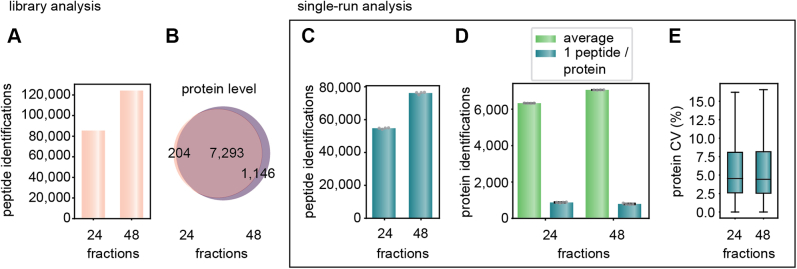
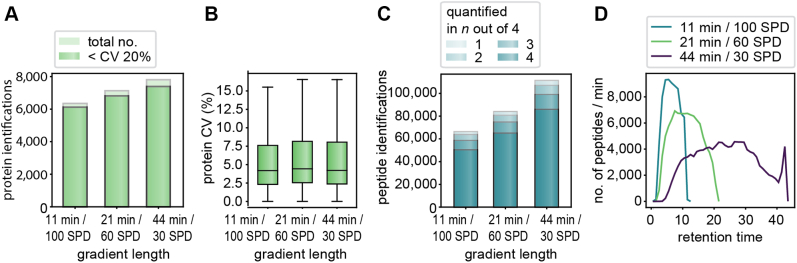
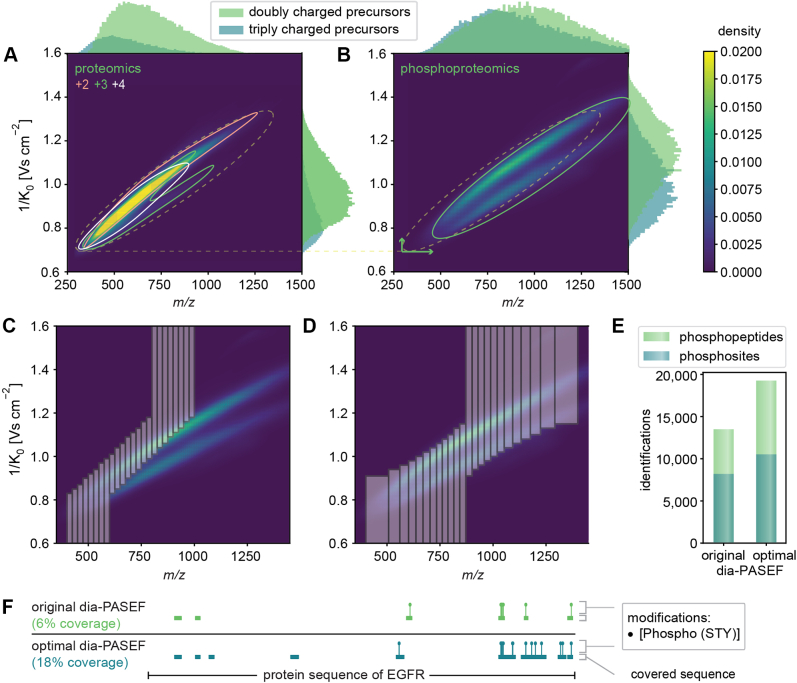
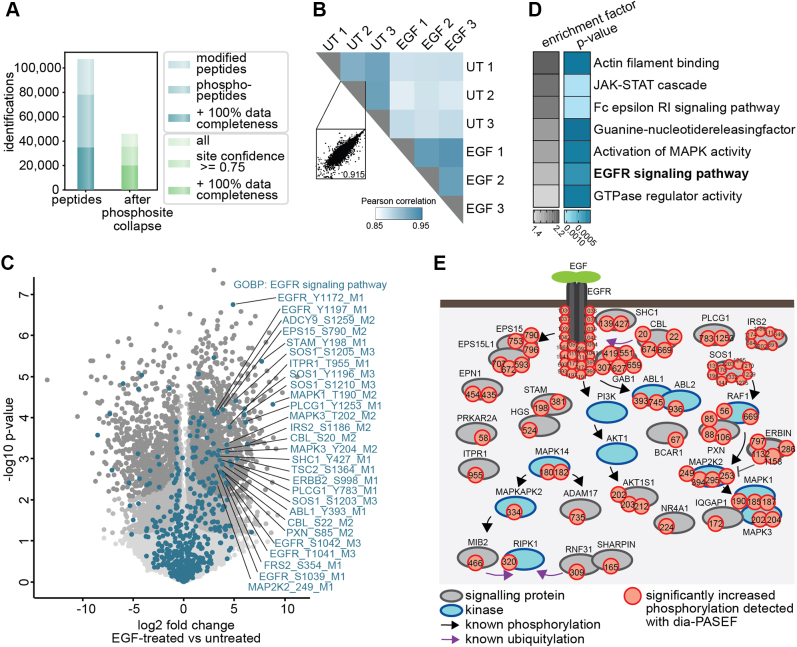
Data-independent acquisition (DIA) methods have become increasingly attractive in mass spectrometry-based proteomics because they enable high data completeness and a wide dynamic range. Recently, we combined DIA with parallel accumulation-serial fragmentation (dia-PASEF) on a Bruker trapped ion mobility (IM) separated quadrupole time-of-flight mass spectrometer. This requires alignment of the IM separation with the downstream mass selective quadrupole, leading to a more complex scheme for dia-PASEF window placement compared with DIA. To achieve high data completeness and deep proteome coverage, here we employ variable isolation windows that are placed optimally depending on precursor density in the m/z and IM plane. This is implemented in the freely available py_diAID (Python package for DIA with an automated isolation design) package. In combination with in-depth project-specific proteomics libraries and the Evosep liquid chromatography system, we reproducibly identified over 7700 proteins in a human cancer cell line in 44 min with quadruplicate single-shot injections at high sensitivity. Even at a throughput of 100 samples per day (11 min liquid chromatography gradients), we consistently quantified more than 6000 proteins in mammalian cell lysates by injecting four replicates. We found that optimal dia-PASEF window placement facilitates in-depth phosphoproteomics with very high sensitivity, quantifying more than 35,000 phosphosites in a human cancer cell line stimulated with an epidermal growth factor in triplicate 21 min runs. This covers a substantial part of the regulated phosphoproteome with high sensitivity, opening up for extensive systems-biological studies.
Keywords: PASEF; TIMS; data-independent acquisition; phosphoproteomics; systems biology.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest M. M. is an indirect investor in Evosep Biosystems. All other authors declare no competing interests.
Figures
References
-
- Aebersold R., Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. - PubMed
-
- Bruderer R., Bernhardt O.M., Gandhi T., Miladinović S.M., Cheng L.Y., Messner S., et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics. 2015;14:1400–1410. - PMC - PubMed
-
- Chapman J.D., Goodlett D.R., Masselon C.D. Multiplexed and data-independent tandem mass spectrometry for global proteome profiling. Mass Spectrom. Rev. 2014;33:452–470. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
