

The mitochondrial NAD kinase functions as a major metabolic regulator upon increased energy demand
- PMID: 35944895
- PMCID: PMC9403569
- DOI: 10.1016/j.molmet.2022.101562
The mitochondrial NAD kinase functions as a major metabolic regulator upon increased energy demand
Abstract
Objective: The mitochondrial nicotinamide adenine dinucleotide (NAD) kinase (MNADK) mediates de novo mitochondrial NADP biosynthesis by catalyzing the phosphorylation of NAD to yield NADP. In this study, we investigated the function and mechanistic basis by which MNADK regulates metabolic homeostasis.
Methods: Generalized gene set analysis by aggregating human patient genomic databases, metabolic studies with genetically engineered animal models, mitochondrial bioenergetic analysis, as well as gain- and loss- of-function studies were performed to address the functions and mechanistic basis by which MNADK regulates energy metabolism and redox state associated with metabolic disease.
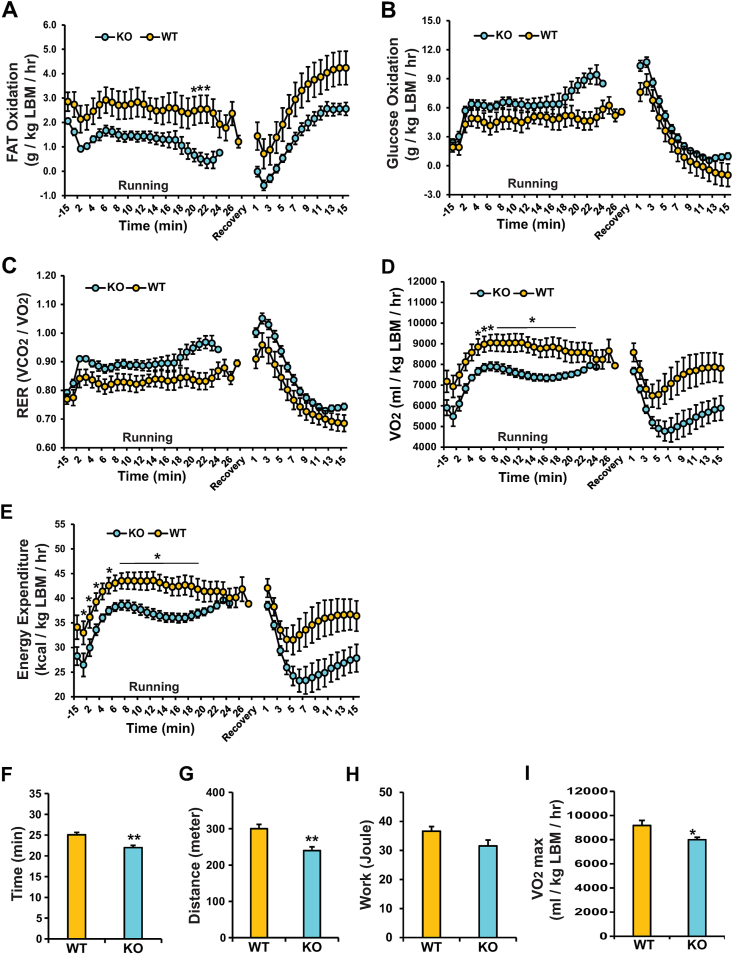
Results: Human MNADK common gene variants or decreased expression of the gene are significantly associated with the occurrence of type-2 diabetes, non-alcoholic fatty liver disease (NAFLD), or hepatocellular carcinoma (HCC). Ablation of the MNADK gene in mice led to decreased fat oxidation, coincident with increased respiratory exchange ratio (RER) and decreased energy expenditure upon energy demand triggered by endurance exercise or fasting. On an atherogenic high-fat diet (HFD), MNADK-null mice exhibited hepatic insulin resistance and glucose intolerance, indicating a type-2 diabetes-like phenotype in the absence of MNADK. MNADK deficiency led to a decrease in mitochondrial NADP(H) but an increase in cellular reactive oxygen species (ROS) in mouse livers. Consistently, protein levels of the major metabolic regulators or enzymes were decreased, while their acetylation modifications were increased in the livers of MNADK-null mice. Feeding mice with a HFD caused S-nitrosylation (SNO) modification, a posttranslational modification that represses protein activities, on MNADK protein in the liver. Reconstitution of an SNO-resistant MNADK variant, MNADK-S193, into MNADK-null mice mitigated hepatic steatosis induced by HFD.
Conclusion: MNADK, the only known mammalian mitochondrial NAD kinase, plays important roles in preserving energy homeostasis to mitigate the risk of metabolic disorders.
Keywords: Acetylation; Diabetes; Energy metabolism; MNADK; Metabolic transcription factors; Mitochondrial metabolism; NADK2.
Copyright © 2022 The Author(s). Published by Elsevier GmbH.. All rights reserved.
Figures








Similar articles
-
Deficiency of the Mitochondrial NAD Kinase Causes Stress-Induced Hepatic Steatosis in Mice.Gastroenterology. 2018 Jan;154(1):224-237. doi: 10.1053/j.gastro.2017.09.010. Epub 2017 Sep 18. Gastroenterology. 2018. PMID: 28923496 Free PMC article.
-
Mitochondrial NAD kinase in health and disease.Redox Biol. 2023 Apr;60:102613. doi: 10.1016/j.redox.2023.102613. Epub 2023 Jan 18. Redox Biol. 2023. PMID: 36689815 Free PMC article.
-
MNADK, a Long-Awaited Human Mitochondrion-Localized NAD Kinase.J Cell Physiol. 2015 Aug;230(8):1697-701. doi: 10.1002/jcp.24926. J Cell Physiol. 2015. PMID: 25641397 Review.
-
MNADK, a novel liver-enriched mitochondrion-localized NAD kinase.Biol Open. 2013 Mar 5;2(4):432-8. doi: 10.1242/bio.20134259. Print 2013 Apr 15. Biol Open. 2013. PMID: 23616928 Free PMC article.
-
Spatiotemporal compartmentalization of hepatic NADH and NADPH metabolism.J Biol Chem. 2018 May 18;293(20):7508-7516. doi: 10.1074/jbc.TM117.000258. Epub 2018 Mar 7. J Biol Chem. 2018. PMID: 29514978 Free PMC article. Review.
Cited by
-
Regulation of and challenges in targeting NAD+ metabolism.Nat Rev Mol Cell Biol. 2024 Oct;25(10):822-840. doi: 10.1038/s41580-024-00752-w. Epub 2024 Jul 18. Nat Rev Mol Cell Biol. 2024. PMID: 39026037 Review.
-
ER-tethered stress sensor CREBH regulates mitochondrial unfolded protein response to maintain energy homeostasis.Proc Natl Acad Sci U S A. 2024 Dec 3;121(49):e2410486121. doi: 10.1073/pnas.2410486121. Epub 2024 Nov 26. Proc Natl Acad Sci U S A. 2024. PMID: 39589874 Free PMC article.
-
Pathobiochemistry of Aging and Neurodegeneration: Deregulation of NAD+ Metabolism in Brain Cells.Biomolecules. 2024 Dec 6;14(12):1556. doi: 10.3390/biom14121556. Biomolecules. 2024. PMID: 39766263 Free PMC article. Review.
-
Homeostatic regulation of NAD(H) and NADP(H) in cells.Genes Dis. 2023 Oct 17;11(5):101146. doi: 10.1016/j.gendis.2023.101146. eCollection 2024 Sep. Genes Dis. 2023. PMID: 38988322 Free PMC article. Review.
-
NADPH Dynamics: Linking Insulin Resistance and β-Cells Ferroptosis in Diabetes Mellitus.Int J Mol Sci. 2023 Dec 26;25(1):342. doi: 10.3390/ijms25010342. Int J Mol Sci. 2023. PMID: 38203517 Free PMC article. Review.
References
-
- Imai S., Armstrong C.M., Kaeberlein M., Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403(6771):795–800. - PubMed
-
- Houten S.M., Denis S., Te Brinke H., Jongejan A., van Kampen A.H., Bradley E.J., et al. Mitochondrial NADP(H) deficiency due to a mutation in NADK2 causes dienoyl-CoA reductase deficiency with hyperlysinemia. Human Molecular Genetics. 2014;23(18):5009–5016. - PubMed
-
- Pollak N., Niere M., Ziegler M. NAD kinase levels control the NADPH concentration in human cells. Journal of Biological Chemistry. 2007;282(46):33562–33571. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
