

Comparison of ONT and CCS sequencing technologies on the polyploid genome of a medicinal plant showed that high error rate of ONT reads are not suitable for self-correction
- PMID: 35945546
- PMCID: PMC9364492
- DOI: 10.1186/s13020-022-00644-1
Comparison of ONT and CCS sequencing technologies on the polyploid genome of a medicinal plant showed that high error rate of ONT reads are not suitable for self-correction
Abstract
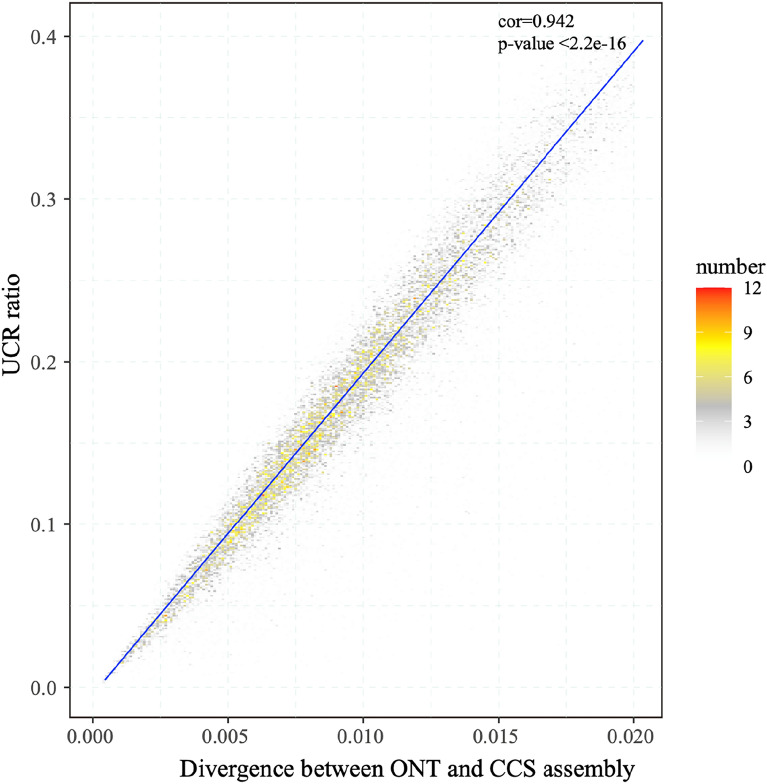
Background: Many medicinal plants are known for their complex genomes with high ploidy, heterozygosity, and repetitive content which pose severe challenges for genome sequencing of those species. Long reads from Oxford nanopore sequencing technology (ONT) or Pacific Biosciences Single Molecule, Real-Time (SMRT) sequencing offer great advantages in de novo genome assembly, especially for complex genomes with high heterozygosity and repetitive content. Currently, multiple allotetraploid species have sequenced their genomes by long-read sequencing. However, we found that a considerable proportion of these genomes (7.9% on average, maximum 23.7%) could not be covered by NGS (Next Generation Sequencing) reads (uncovered region by NGS reads, UCR) suggesting the questionable and low-quality of those area or genomic areas that can't be sequenced by NGS due to sequencing bias. The underlying causes of those UCR in the genome assembly and solutions to this problem have never been studied.
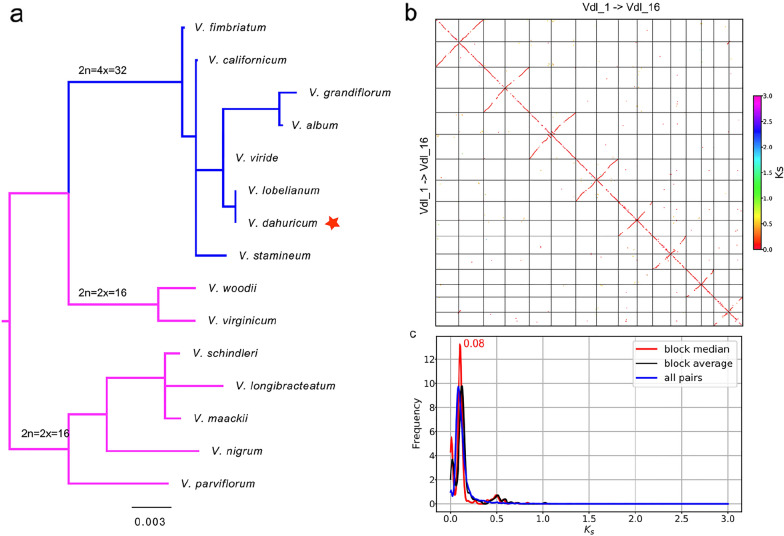
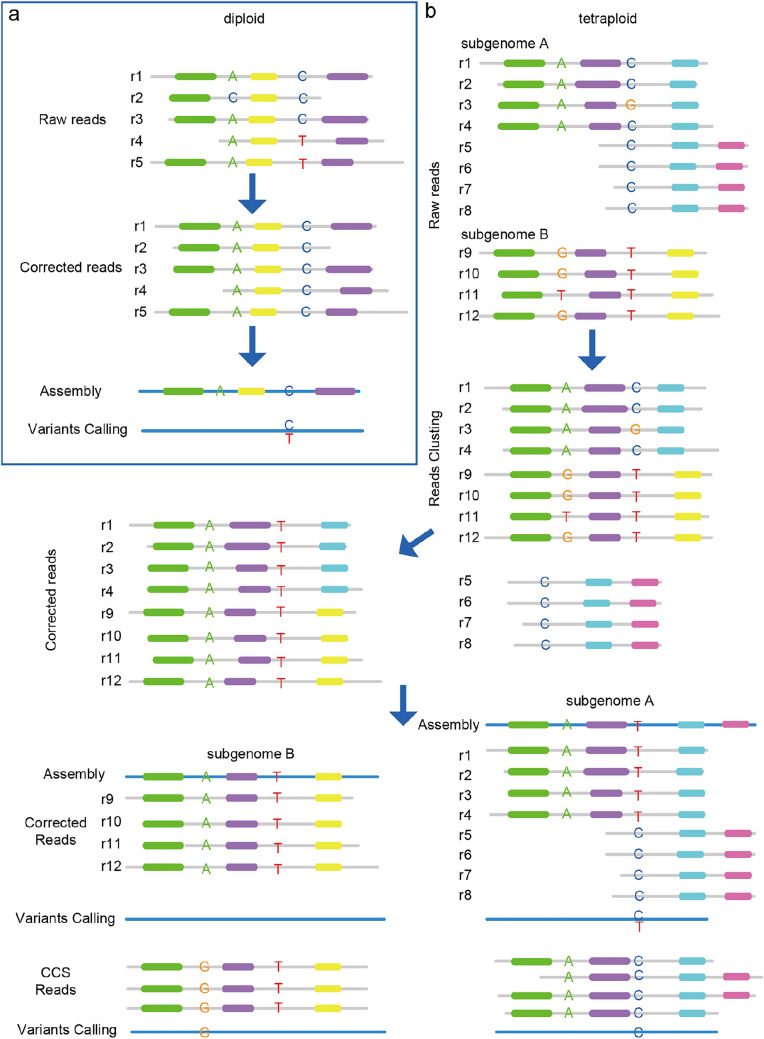
Methods: In the study, we sequenced the tetraploid genome of Veratrum dahuricum (Turcz.) O. Loes (VDL), a Chinese medicinal plant, with ONT platform and assembled the genome with three strategies in parallel. We compared the qualities, coverage, and heterozygosity of the three ONT assemblies with another released assembly of the same individual using reads from PacBio circular consensus sequencing (CCS) technology, to explore the cause of the UCR.
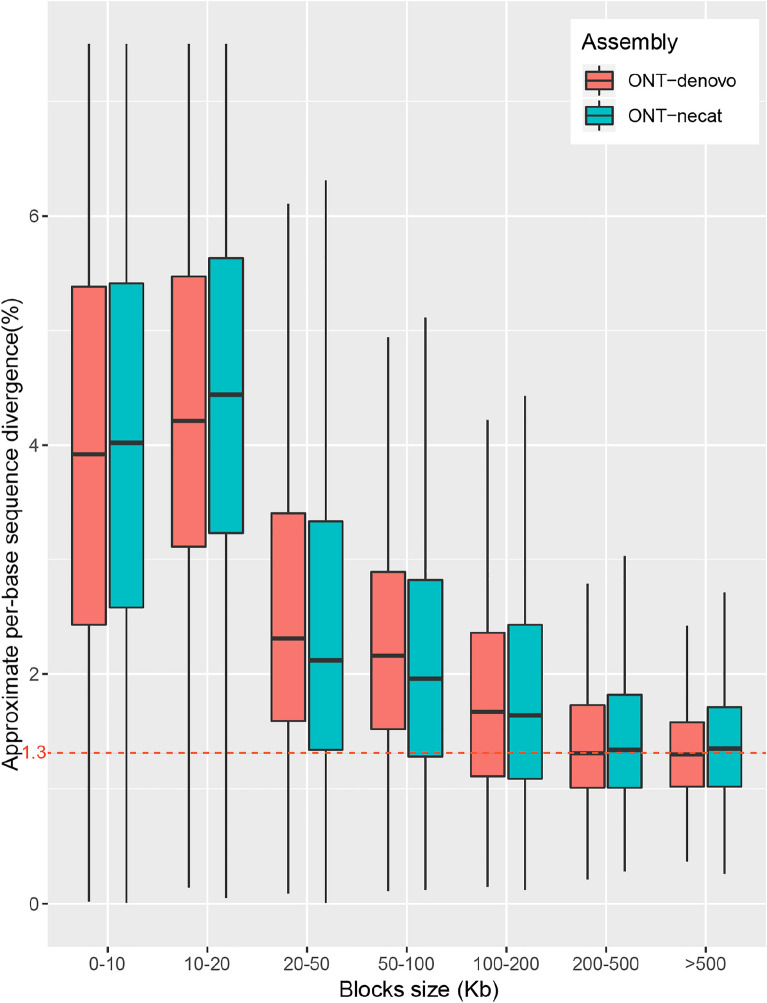
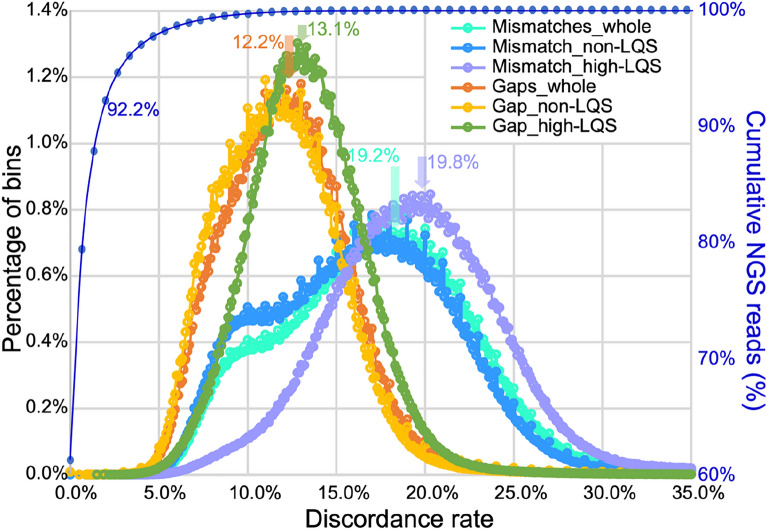
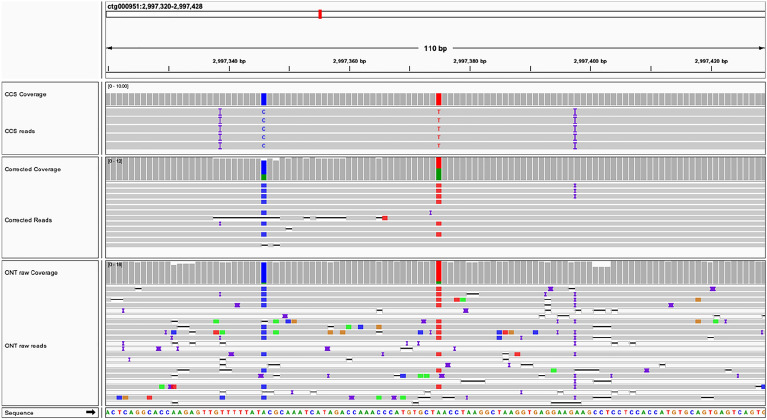
Results: By mapping the NGS reads against the three ONT assemblies and the CCS assembly, we found that the coverage of those ONT assemblies by NGS reads ranged from 49.15 to 76.31%, much smaller than that of the CCS assembly (99.53%). And alignment between ONT assemblies and CCS assembly showed that most UCR can be aligned with CCS assembly. So, we conclude that the UCRs in ONT assembly are low-quality sequences with a high error rate that can't be aligned with short reads, rather than genomic regions that can't be sequenced by NGS. Further comparison among the intermediate versions of ONT assemblies showed that the most probable origin of those errors is a combination of artificial errors introduced by "self-correction" and initial sequencing error in long reads. We also found that polishing the ONT assembly with CCS reads can correct those errors efficiently.
Conclusions: Through analyzing genome features and reads alignment, we have found the causes for the high proportion of UCR in ONT assembly of VDL are sequencing errors and additional errors introduced by self-correction. The high error rates of ONT-raw reads make them not suitable for self-correction prior to allotetraploid genome assembly, as the self-correction will introduce artificial errors to > 5% of the UCR sequences. We suggest high-precision CCS reads be used to polish the assembly to correct those errors effectively for polyploid genomes.
Keywords: Allotetraploid; Homozygous variants; Low-quality sequences; ONT-based assembly; Veratrum dahuricum.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
Grants and funding
- 031/2017/A1/The Science and Technology Development Fund Macau SAR
- 3102019JC007/the Talents Team Construction Fund of Northwestern Polytechnical University (NWPU), the Fundamental Research Funds for the Central Universities
- 5113190037/Ten Thousand Talent Plans for Young Top-notch Talents of Yunnan Province
LinkOut - more resources
Full Text Sources
