

Loss of heat shock factor 1 promotes hepatic stellate cell activation and drives liver fibrosis
- PMID: 35945902
- PMCID: PMC9512451
- DOI: 10.1002/hep4.2058
Loss of heat shock factor 1 promotes hepatic stellate cell activation and drives liver fibrosis
Abstract
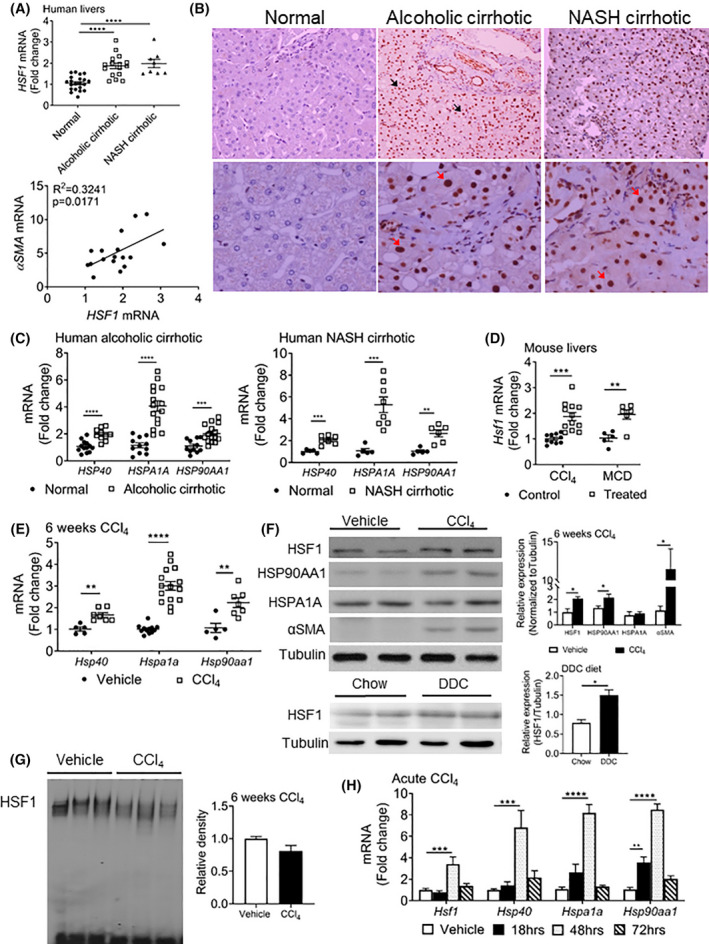
Liver fibrosis is an aberrant wound healing response that results from chronic injury and is mediated by hepatocellular death and activation of hepatic stellate cells (HSCs). While induction of oxidative stress is well established in fibrotic livers, there is limited information on stress-mediated mechanisms of HSC activation. Cellular stress triggers an adaptive defense mechanism via master protein homeostasis regulator, heat shock factor 1 (HSF1), which induces heat shock proteins to respond to proteotoxic stress. Although the importance of HSF1 in restoring cellular homeostasis is well-established, its potential role in liver fibrosis is unknown. Here, we show that HSF1 messenger RNA is induced in human cirrhotic and murine fibrotic livers. Hepatocytes exhibit nuclear HSF1, whereas stellate cells expressing alpha smooth muscle actin do not express nuclear HSF1 in human cirrhosis. Interestingly, despite nuclear HSF1, murine fibrotic livers did not show induction of HSF1 DNA binding activity compared with controls. HSF1-deficient mice exhibit augmented HSC activation and fibrosis despite limited pro-inflammatory cytokine response and display delayed fibrosis resolution. Stellate cell and hepatocyte-specific HSF1 knockout mice exhibit higher induction of profibrogenic response, suggesting an important role for HSF1 in HSC activation and fibrosis. Stable expression of dominant negative HSF1 promotes fibrogenic activation of HSCs. Overactivation of HSF1 decreased phosphorylation of JNK and prevented HSC activation, supporting a protective role for HSF1. Our findings identify an unconventional role for HSF1 in liver fibrosis. Conclusion: Our results show that deficiency of HSF1 is associated with exacerbated HSC activation promoting liver fibrosis, whereas activation of HSF1 prevents profibrogenic HSC activation.
© 2022 The Authors. Hepatology Communications published by Wiley Periodicals LLC on behalf of American Association for the Study of Liver Diseases.
Conflict of interest statement
Nothing to report.
Figures







Similar articles
-
Coordinated signaling of activating transcription factor 6α and inositol-requiring enzyme 1α regulates hepatic stellate cell-mediated fibrogenesis in mice.Am J Physiol Gastrointest Liver Physiol. 2021 May 1;320(5):G864-G879. doi: 10.1152/ajpgi.00453.2020. Epub 2021 Mar 17. Am J Physiol Gastrointest Liver Physiol. 2021. PMID: 33728997 Free PMC article.
-
Inhibition of protein arginine methyltransferase 1 alleviates liver fibrosis by attenuating the activation of hepatic stellate cells in mice.FASEB J. 2022 Sep;36(9):e22489. doi: 10.1096/fj.202200238R. FASEB J. 2022. PMID: 35959865
-
The HLF/IL-6/STAT3 feedforward circuit drives hepatic stellate cell activation to promote liver fibrosis.Gut. 2018 Sep;67(9):1704-1715. doi: 10.1136/gutjnl-2016-313392. Epub 2017 Jul 28. Gut. 2018. PMID: 28754776
-
Liver Fibrosis: From Basic Science towards Clinical Progress, Focusing on the Central Role of Hepatic Stellate Cells.Int J Mol Sci. 2024 Jul 18;25(14):7873. doi: 10.3390/ijms25147873. Int J Mol Sci. 2024. PMID: 39063116 Free PMC article. Review.
-
Cooperation of liver cells in health and disease.Adv Anat Embryol Cell Biol. 2001;161:III-XIII, 1-151. doi: 10.1007/978-3-642-56553-3. Adv Anat Embryol Cell Biol. 2001. PMID: 11729749 Review.
Cited by
-
Targeting prominin-2/BACH1/GLS pathway to inhibit oxidative stress-induced ferroptosis of bone mesenchymal stem cells.Stem Cell Res Ther. 2025 Apr 29;16(1):213. doi: 10.1186/s13287-025-04326-1. Stem Cell Res Ther. 2025. PMID: 40301995 Free PMC article.
-
A computational framework to in silico screen for drug-induced hepatocellular toxicity.Toxicol Sci. 2024 Sep 1;201(1):14-25. doi: 10.1093/toxsci/kfae078. Toxicol Sci. 2024. PMID: 38902949 Free PMC article.
-
The role of JNK signaling pathway in organ fibrosis.J Adv Res. 2025 Aug;74:207-223. doi: 10.1016/j.jare.2024.09.029. Epub 2024 Oct 2. J Adv Res. 2025. PMID: 39366483 Free PMC article. Review.
References
-
- Yoon YH, Chen CM. Liver cirrhosis mortality in the United States: national, state, and regional trends, 2000–2015 National Institute on Alcohol Abuse and Alcoholism Division of Epidemiology and Prevention Research Alcohol Epidemiologic Data System 2018. Surveillance Report. 2018;111:1–73.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
