

Analysis of co-occurring and mutually exclusive amino acid changes and detection of convergent and divergent evolution events in SARS-CoV-2
- PMID: 35945925
- PMCID: PMC9352683
- DOI: 10.1016/j.csbj.2022.07.051
Analysis of co-occurring and mutually exclusive amino acid changes and detection of convergent and divergent evolution events in SARS-CoV-2
Abstract
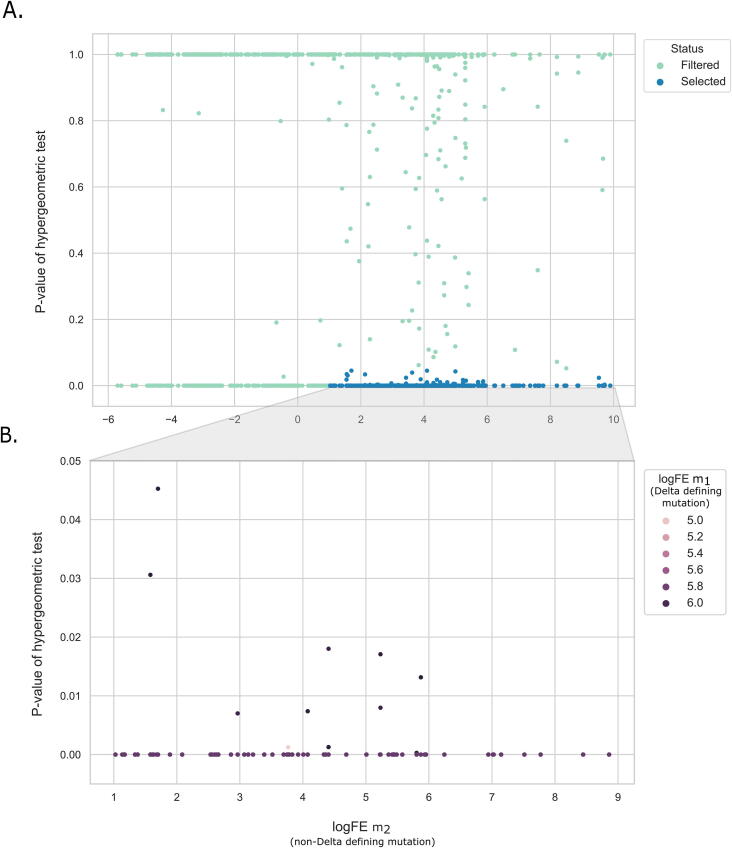
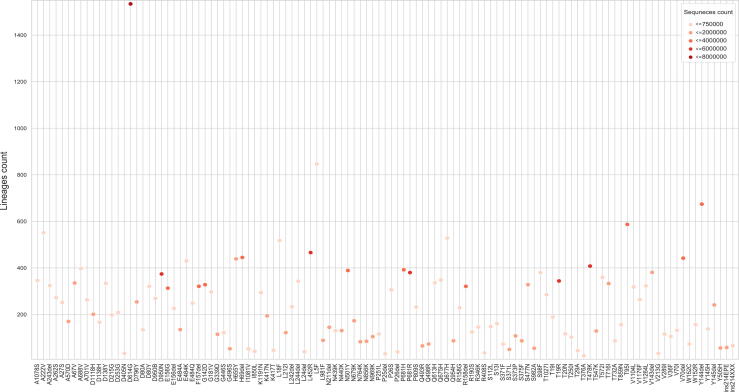
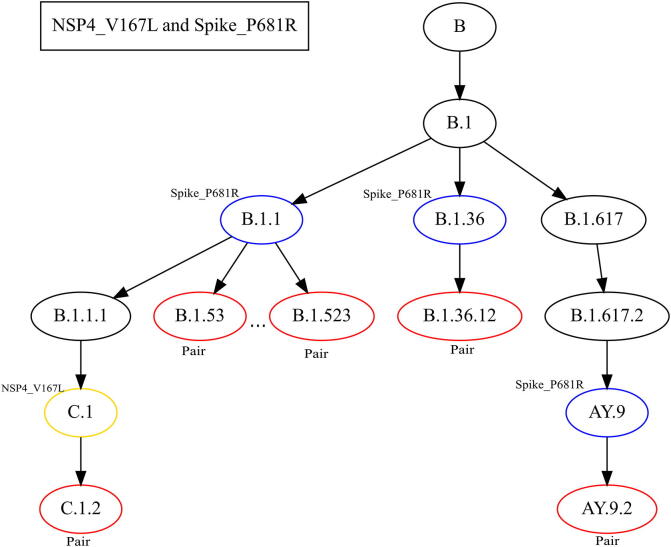
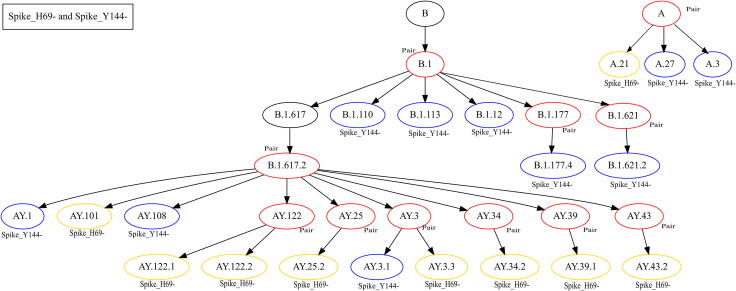
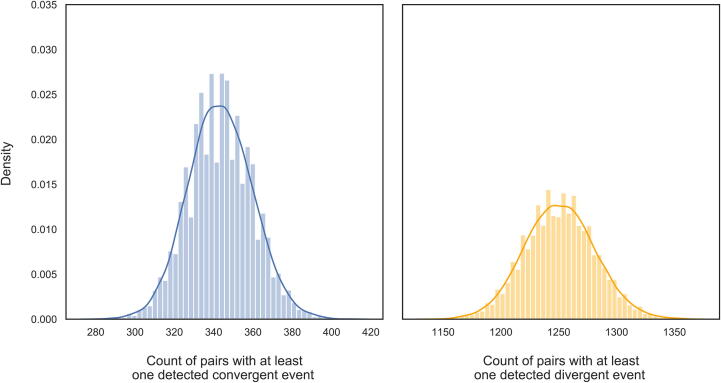
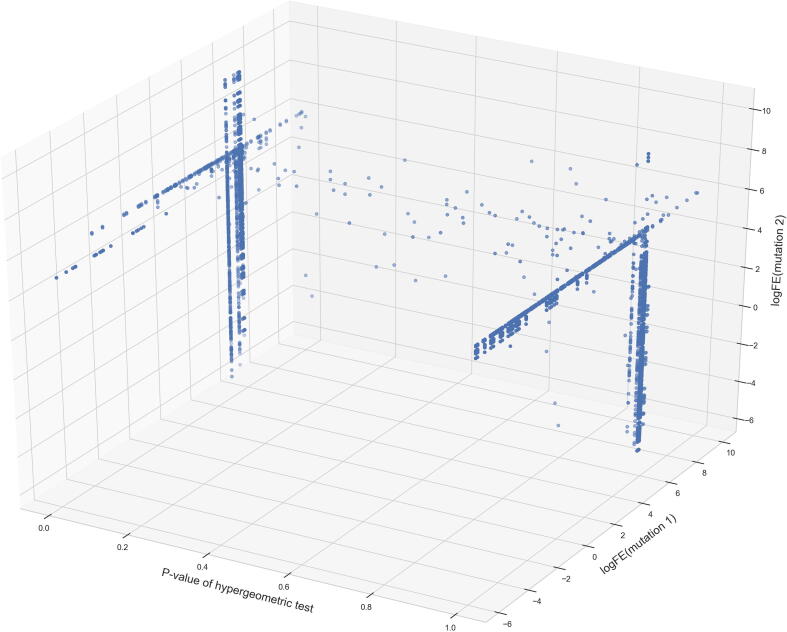
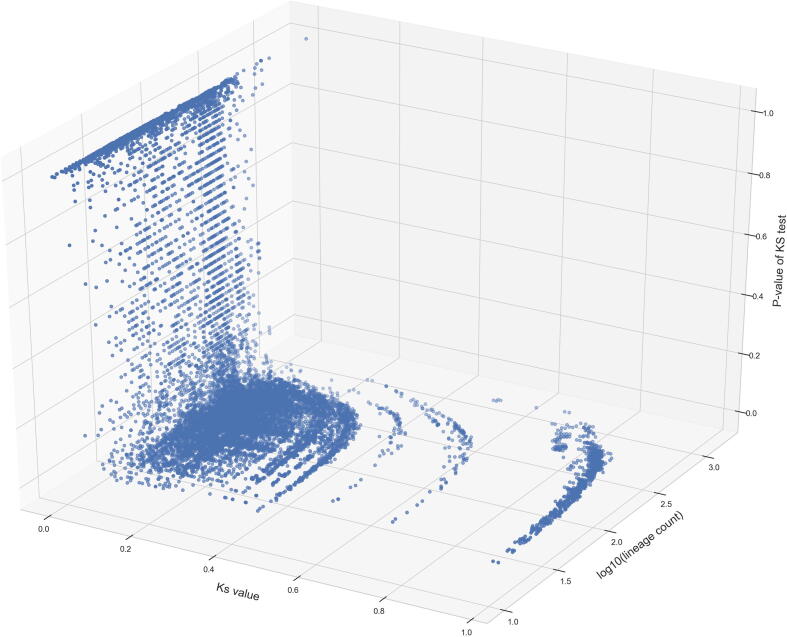
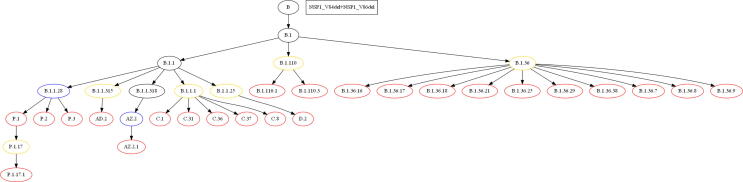
The inflation of SARS-CoV-2 lineages with a high number of accumulated mutations (such as the recent case of Omicron) has risen concerns about the evolutionary capacity of this virus. Here, we propose a computational study to examine non-synonymous mutations gathered within genomes of SARS-CoV-2 from the beginning of the pandemic until February 2022. We provide both qualitative and quantitative descriptions of such corpus, focusing on statistically significant co-occurring and mutually exclusive mutations within single genomes. Then, we examine in depth the distributions of mutations over defined lineages and compare those of frequently co-occurring mutation pairs. Based on this comparison, we study mutations' convergence/divergence on the phylogenetic tree. As a result, we identify 1,818 co-occurring pairs of non-synonymous mutations showing at least one event of convergent evolution and 6,625 co-occurring pairs with at least one event of divergent evolution. Notable examples of both types are shown by means of a tree-based representation of lineages, visually capturing mutations' behaviors. Our method confirms several well-known cases; moreover, the provided evidence suggests that our workflow can explain aspects of the future mutational evolution of SARS-CoV-2.
Keywords: Co-occurring mutations; Convergent evolution; Divergent evolution; Mutually exclusive mutations; SARS-CoV-2; Statistical testing.
© 2022 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
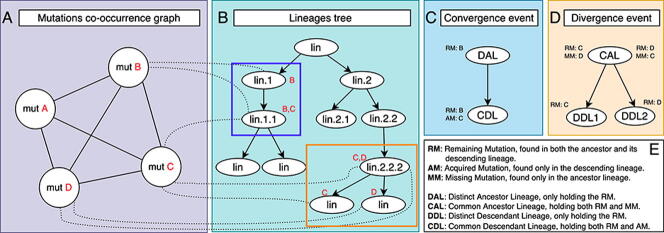
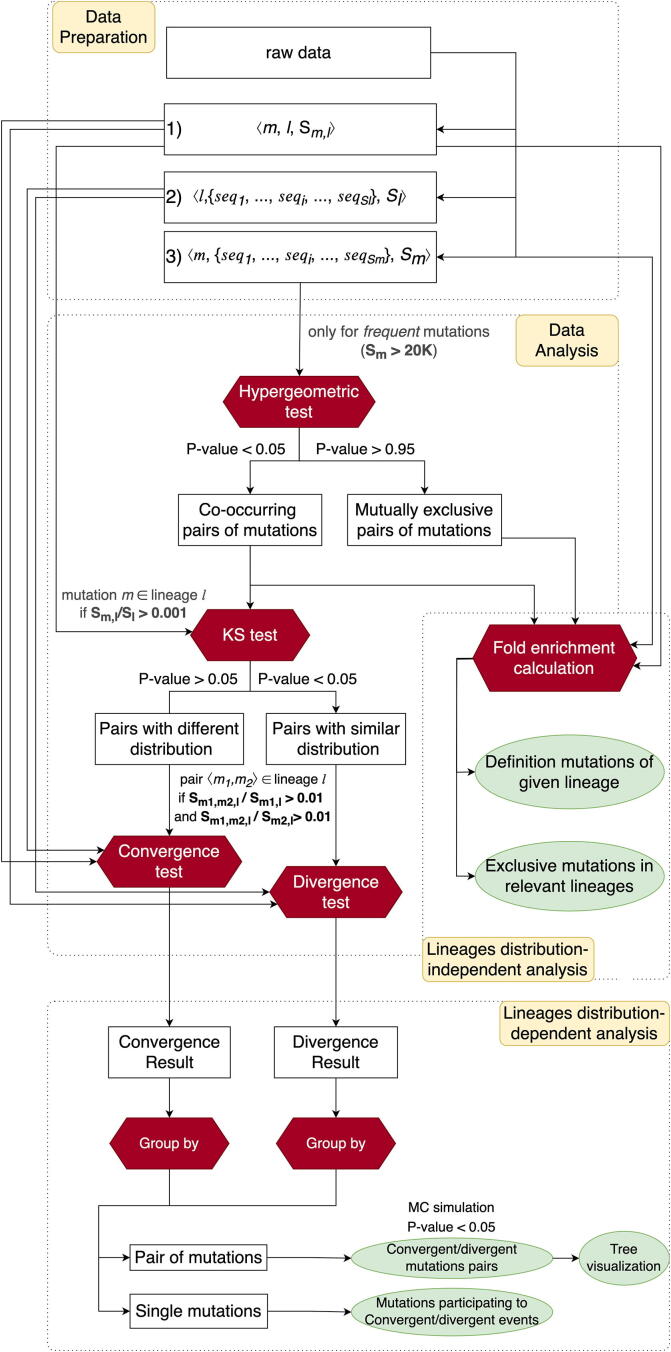
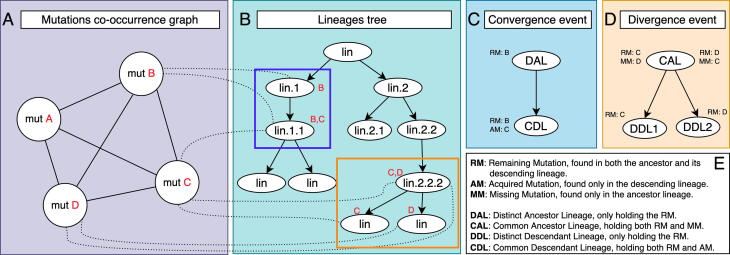
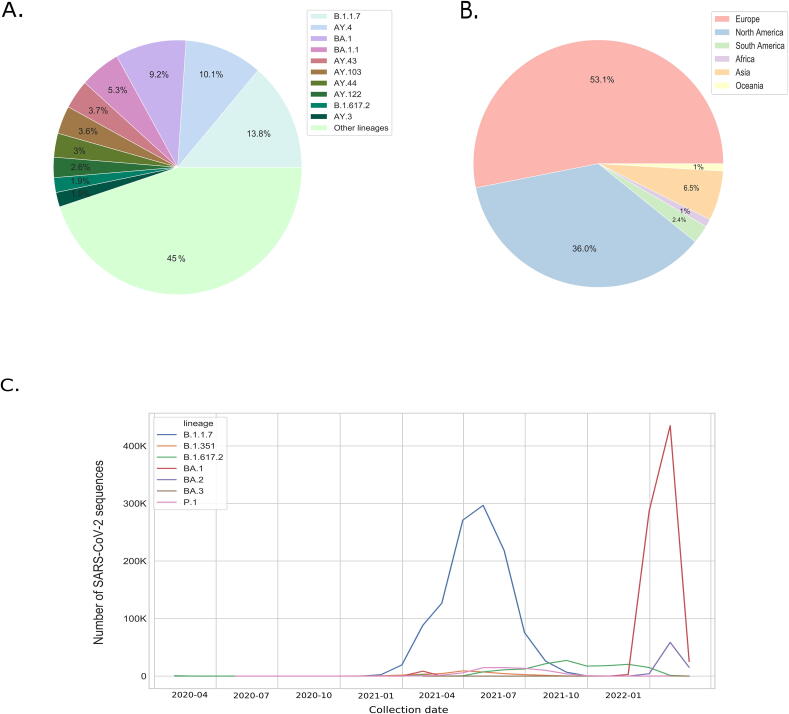
References
-
- Al Khalaf R., Alfonsi T., Ceri S., Bernasconi A. CoV2K: a knowledge base of SARS-CoV-2 variant impacts. International Conference on Research Challenges in Information Science, Springer. 2021:274–282.
-
- Andreano E., Rappuoli R. SARS-CoV-2 escaped natural immunity, raising questions about vaccines and therapies. Nat Med. 2021;27:759–761. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous