

Uncovering protein function: from classification to complexes
- PMID: 35946411
- PMCID: PMC9400073
- DOI: 10.1042/EBC20200108
Uncovering protein function: from classification to complexes
Abstract
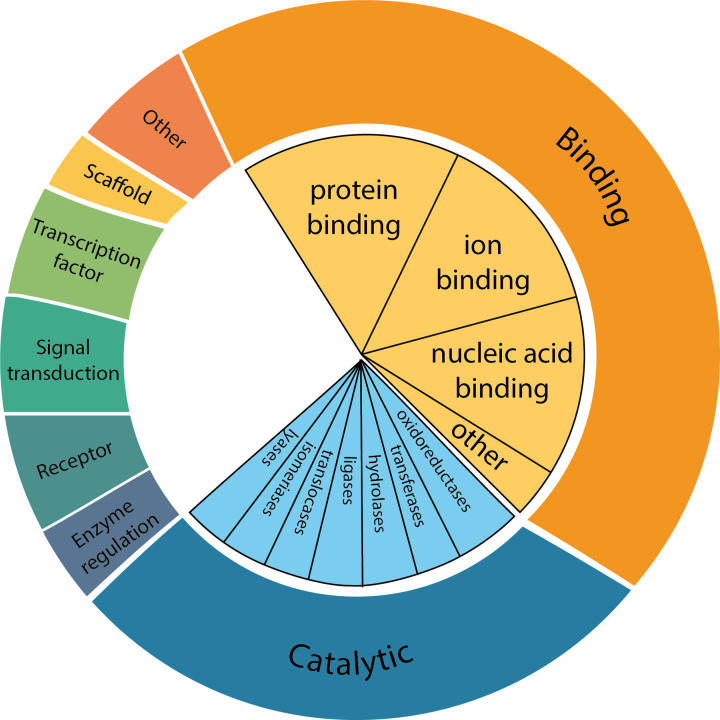
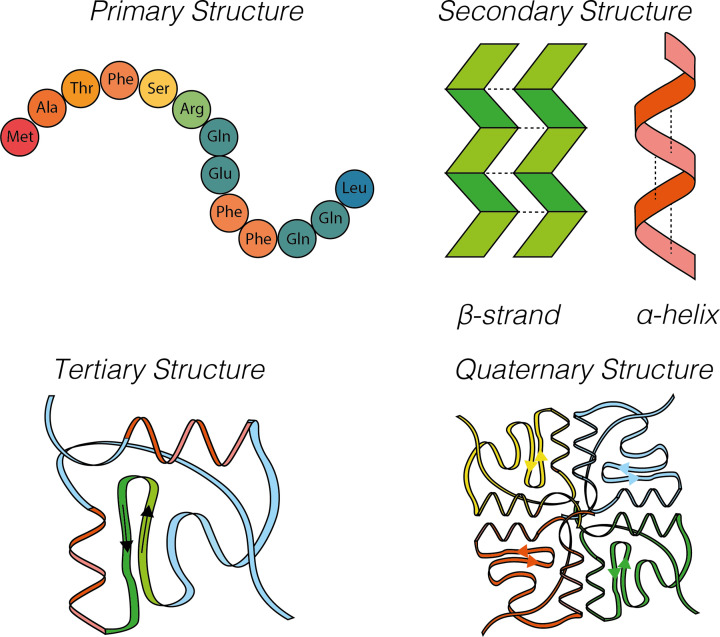
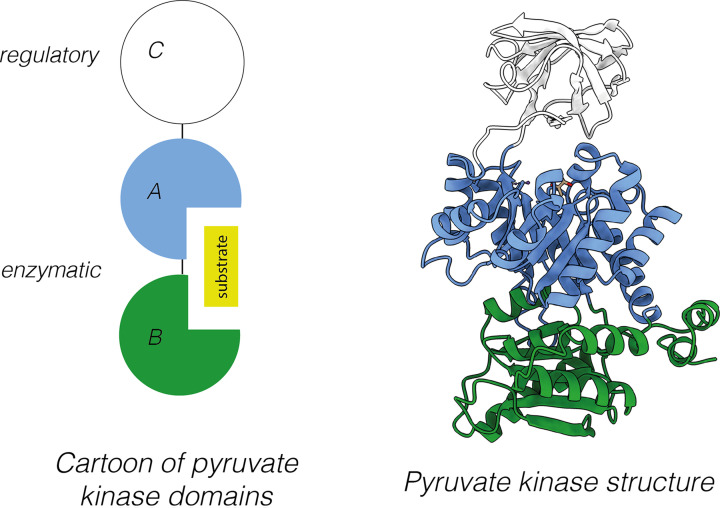
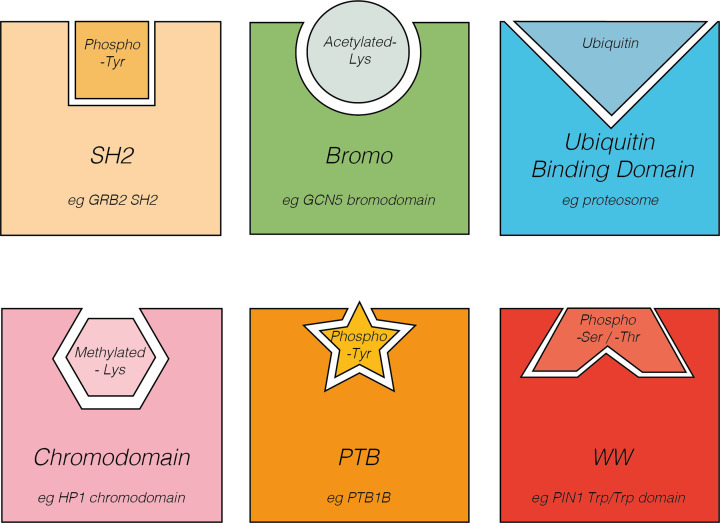
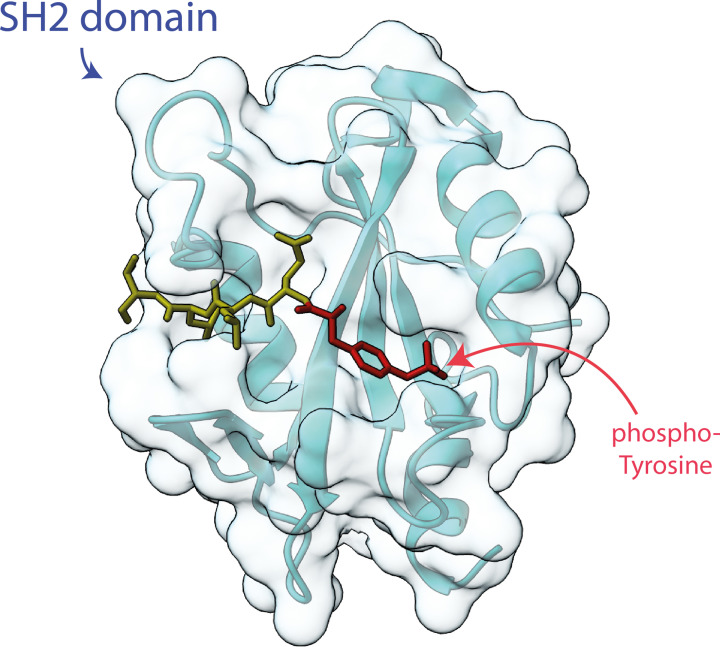
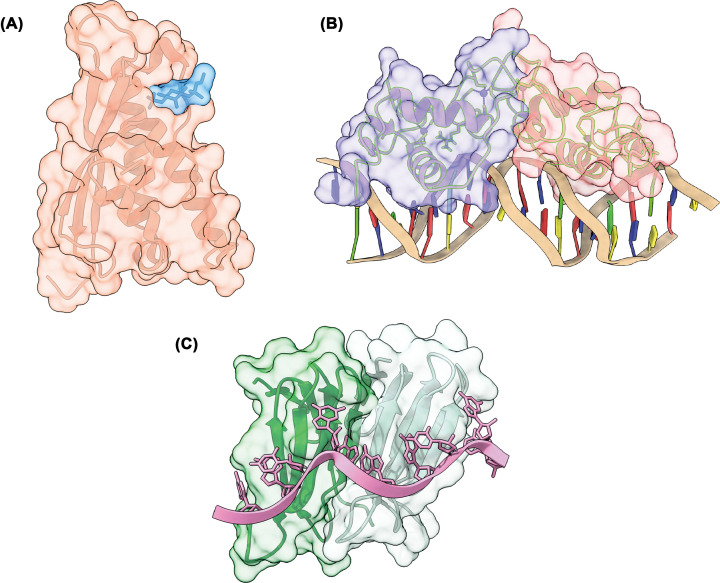
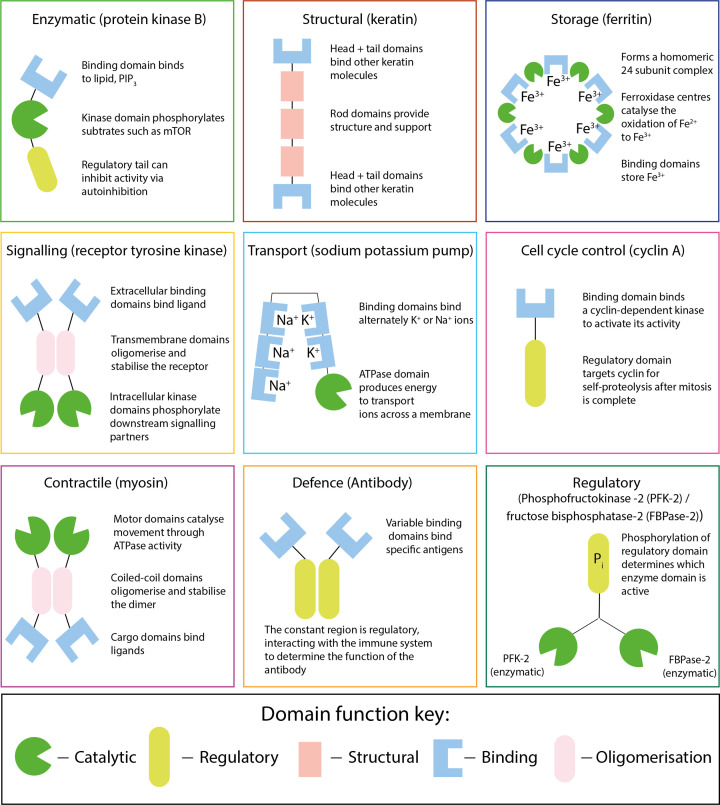
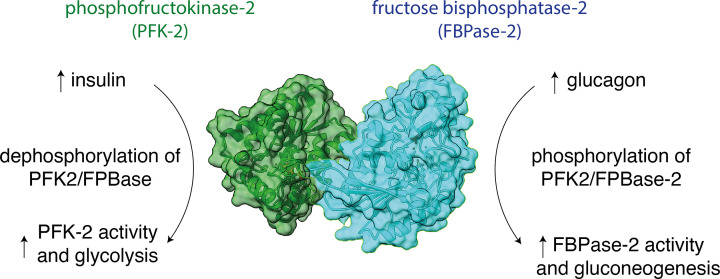
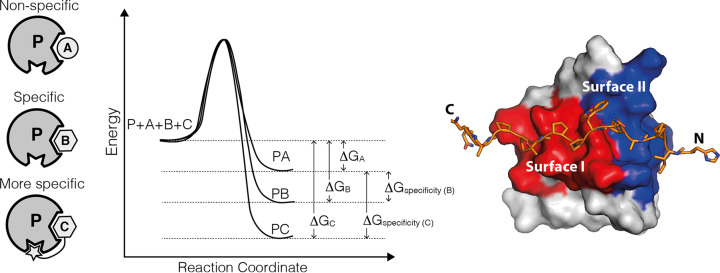
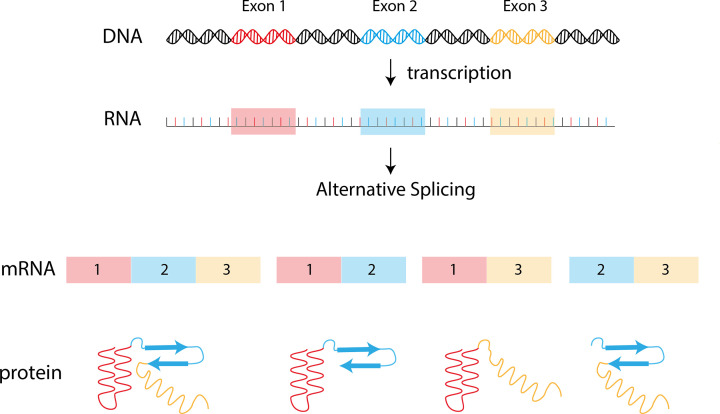
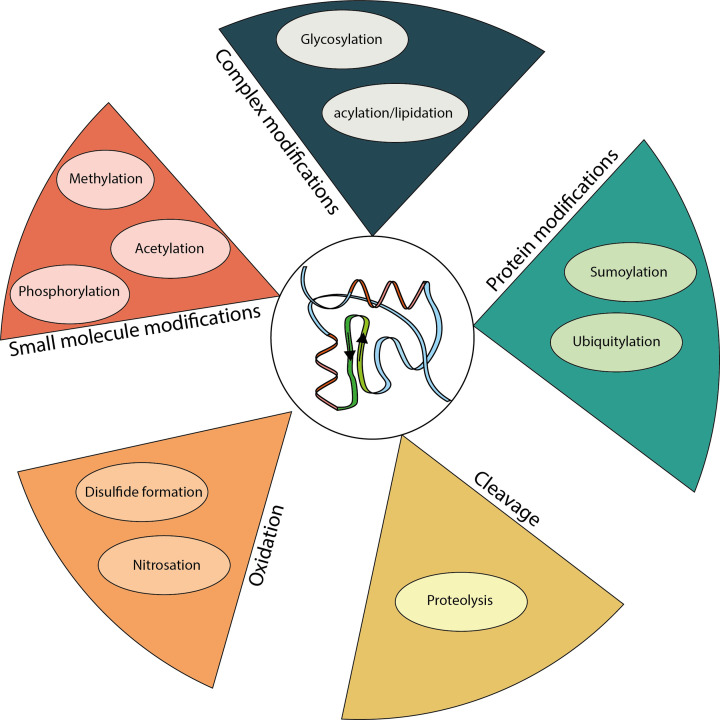
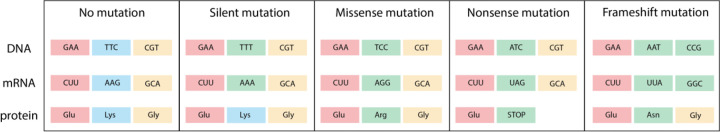
Almost all interactions and reactions that occur in living organisms involve proteins. The various biological roles of proteins include, but are not limited to, signal transduction, gene transcription, cell death, immune function, structural support, and catalysis of all the chemical reactions that enable organisms to survive. The varied roles of proteins have led to them being dubbed 'the workhorses of all living organisms'. This article discusses the functions of proteins and how protein function is studied in a laboratory setting. In this article, we begin by examining the functions of protein domains, followed by a discussion of some of the major classes of proteins based on their function. We consider protein binding in detail, which is central to protein function. We then examine how protein function can be altered through various mechanisms including post-translational modification, and changes to environment, oligomerisation and mutations. Finally, we consider a handful of the techniques employed in the laboratory to understand and measure the function of proteins.
Keywords: biochemical techniques and resources; post translational modification; protein binding.
© 2022 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures
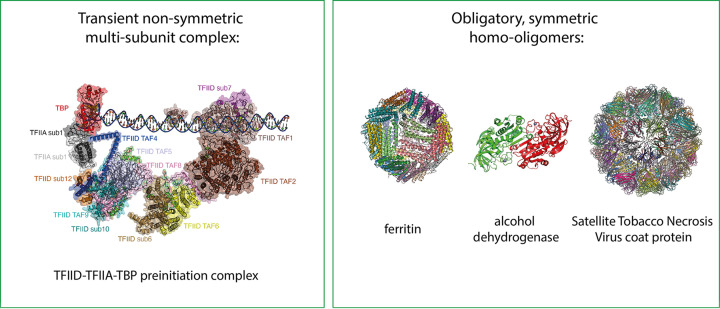
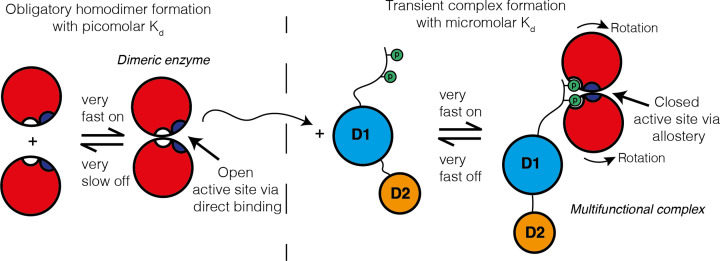
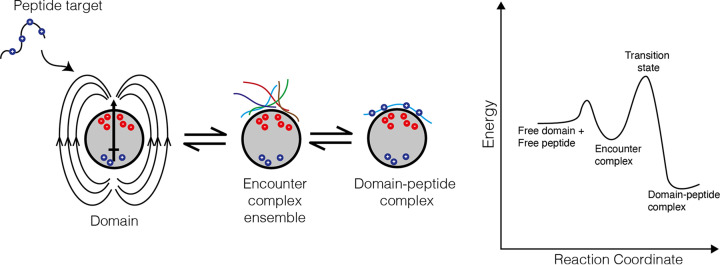
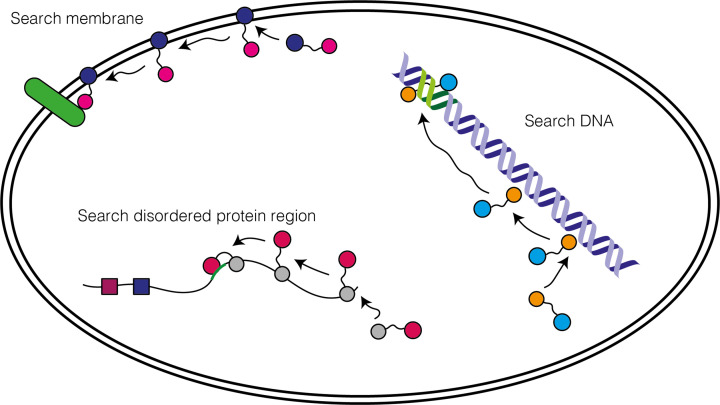
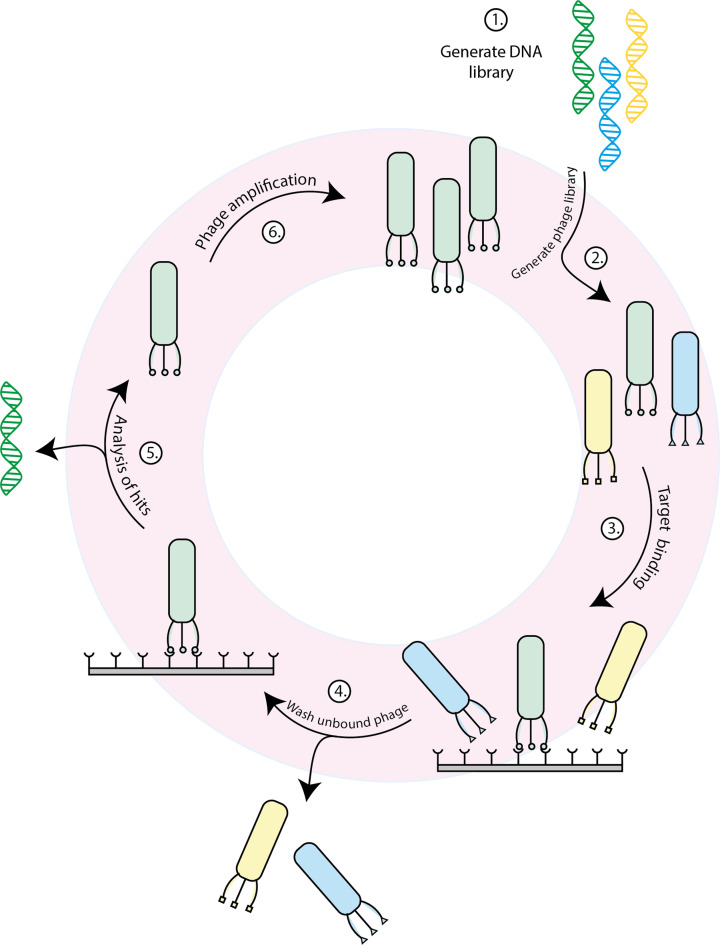
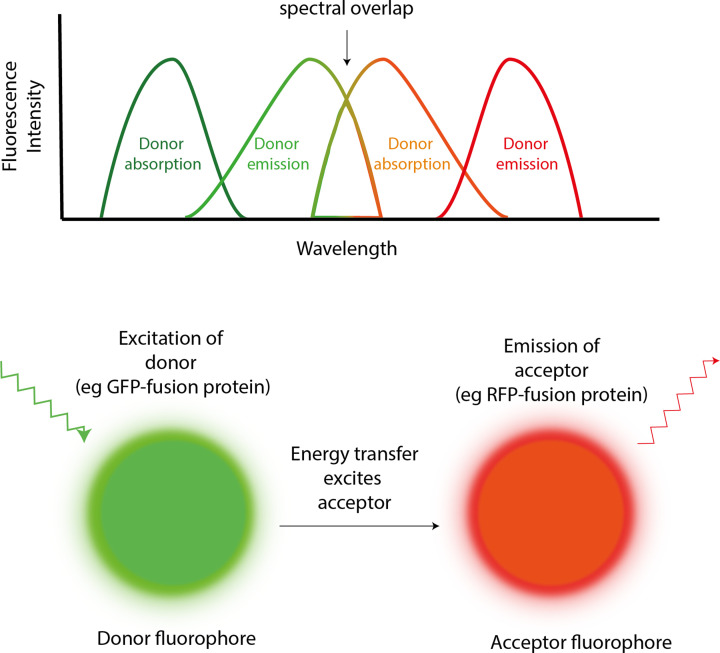
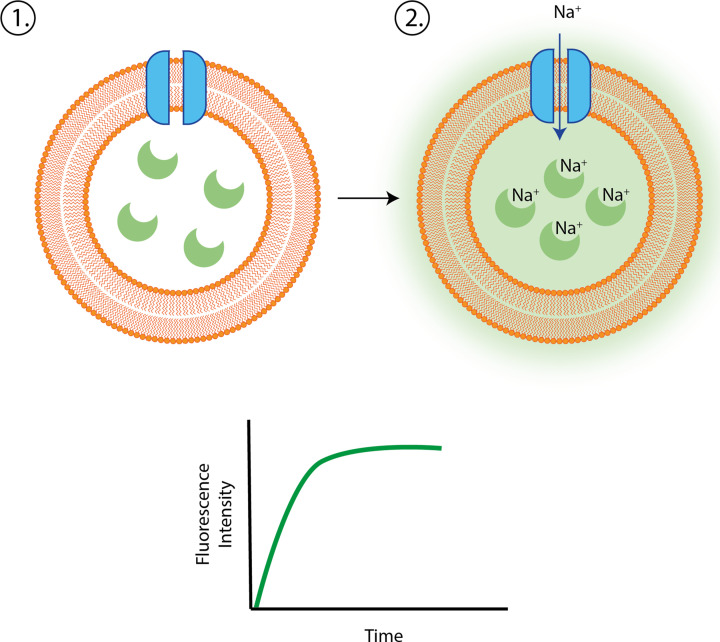
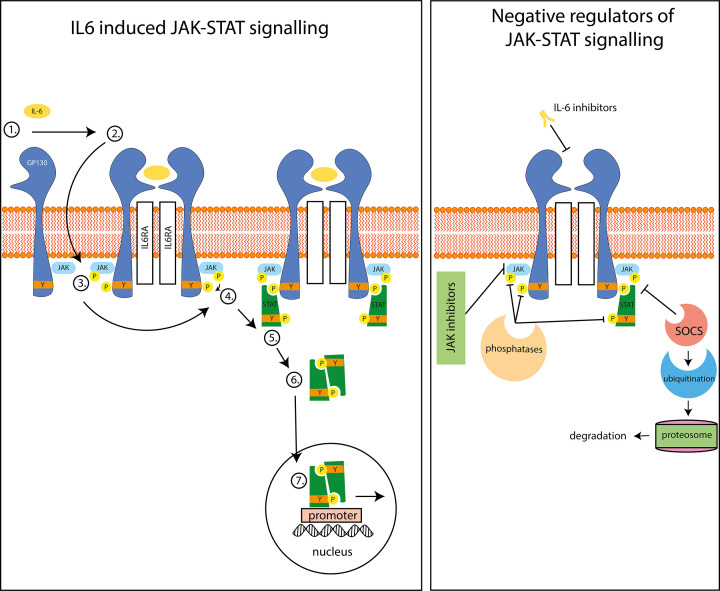
References
-
- Watch a video on proteins and see their range of structures and functions: https://pdb101.rcsb.org/learn/videos/what-is-a-protein-video (Feb, 2022)
-
- Explore protein folding on your computer with the game Foldit: https://fold.it/ (Feb, 2022)
-
- Read about how de novo proteins promise new covid vaccines and medicines: https://www.scientificamerican.com/article/artificial-proteins-never-se... (Feb, 2022)
-
- Look at the predicted structures of proteins in the AlphaFold database: https://alphafold.ebi.ac.uk/ (Feb, 2022)
-
- Learn more about biophysical techniques for measuring protein function: https://portlandpress.com/emergtoplifesci/article-abstract/5/1/1/228110/... (Feb, 2022)
MeSH terms
Substances
LinkOut - more resources
Full Text Sources