

Distinct evolutionary trajectories of SARS-CoV-2-interacting proteins in bats and primates identify important host determinants of COVID-19
- PMID: 35947637
- PMCID: PMC9436378
- DOI: 10.1073/pnas.2206610119
Distinct evolutionary trajectories of SARS-CoV-2-interacting proteins in bats and primates identify important host determinants of COVID-19
Abstract
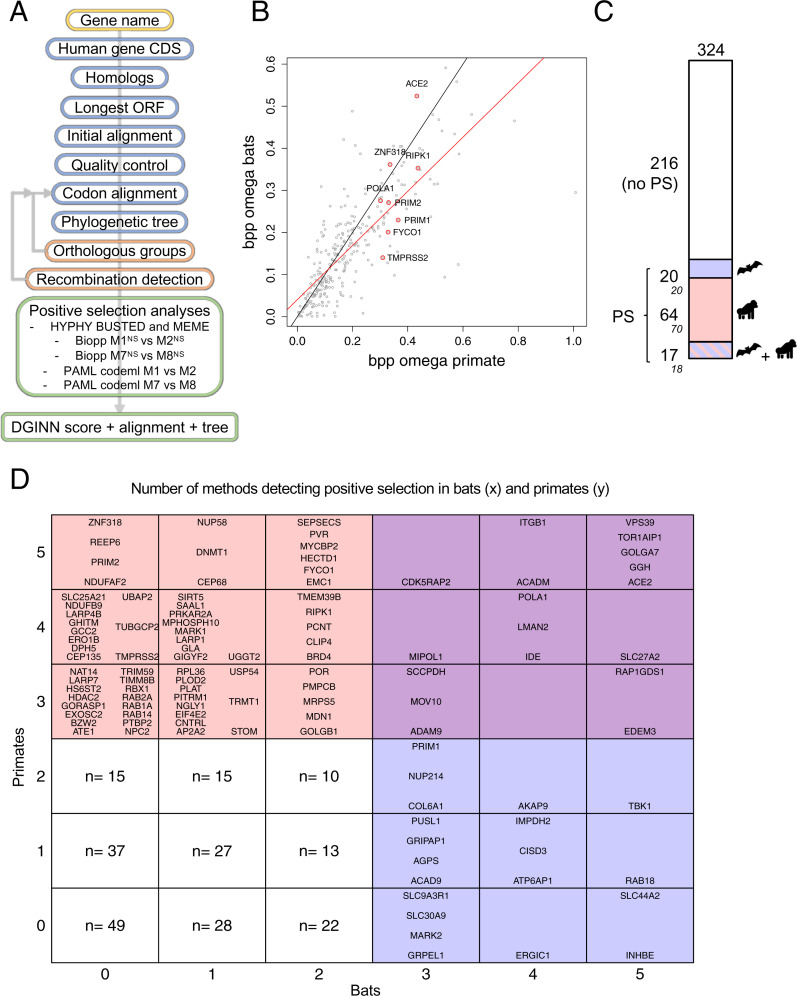
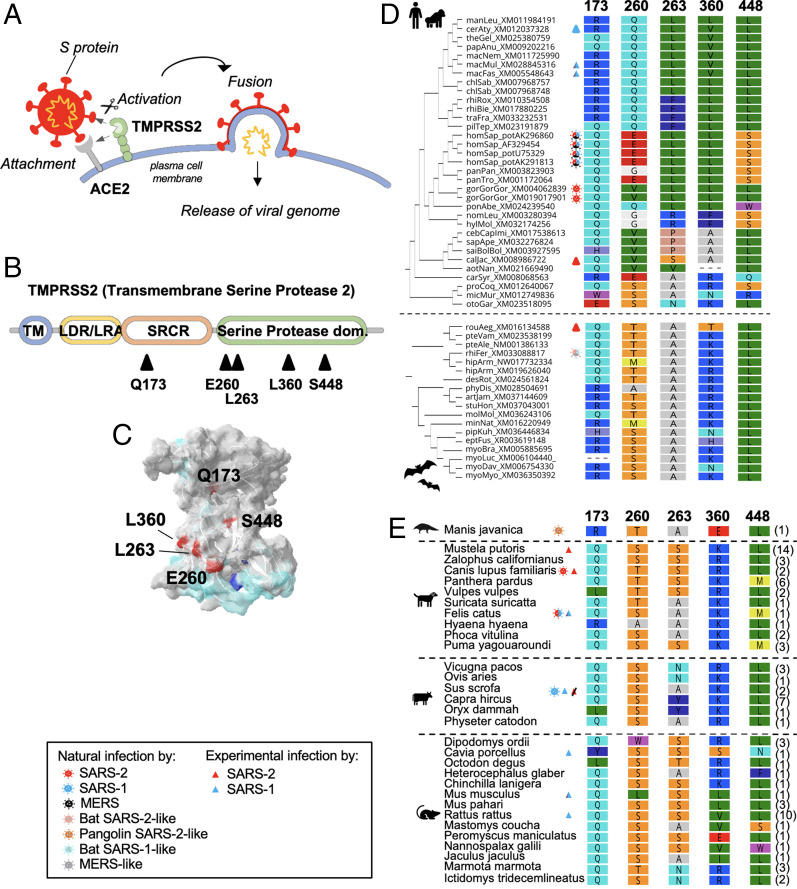
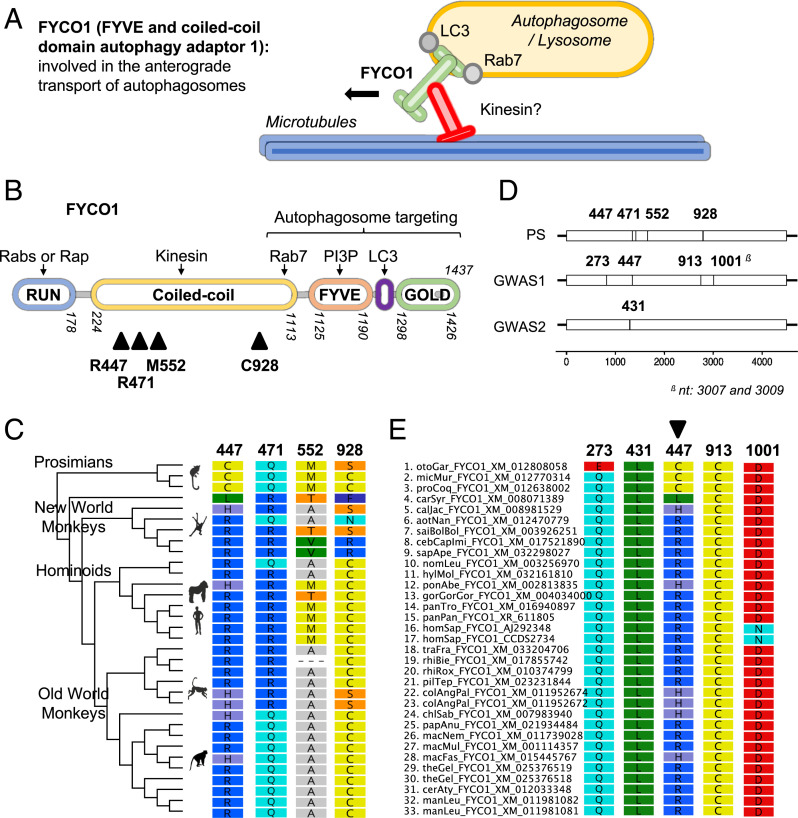
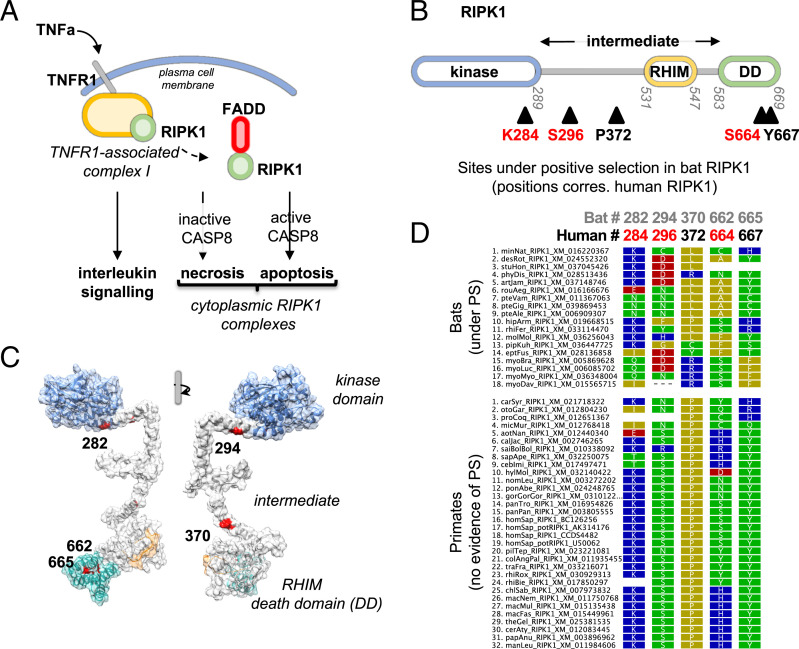
The coronavirus disease 19 (COVID-19) pandemic is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a coronavirus that spilled over from the bat reservoir. Despite numerous clinical trials and vaccines, the burden remains immense, and the host determinants of SARS-CoV-2 susceptibility and COVID-19 severity remain largely unknown. Signatures of positive selection detected by comparative functional genetic analyses in primate and bat genomes can uncover important and specific adaptations that occurred at virus-host interfaces. We performed high-throughput evolutionary analyses of 334 SARS-CoV-2-interacting proteins to identify SARS-CoV adaptive loci and uncover functional differences between modern humans, primates, and bats. Using DGINN (Detection of Genetic INNovation), we identified 38 bat and 81 primate proteins with marks of positive selection. Seventeen genes, including the ACE2 receptor, present adaptive marks in both mammalian orders, suggesting common virus-host interfaces and past epidemics of coronaviruses shaping their genomes. Yet, 84 genes presented distinct adaptations in bats and primates. Notably, residues involved in ubiquitination and phosphorylation of the inflammatory RIPK1 have rapidly evolved in bats but not primates, suggesting different inflammation regulation versus humans. Furthermore, we discovered residues with typical virus-host arms race marks in primates, such as in the entry factor TMPRSS2 or the autophagy adaptor FYCO1, pointing to host-specific in vivo interfaces that may be drug targets. Finally, we found that FYCO1 sites under adaptation in primates are those associated with severe COVID-19, supporting their importance in pathogenesis and replication. Overall, we identified adaptations involved in SARS-CoV-2 infection in bats and primates, enlightening modern genetic determinants of virus susceptibility and severity.
Keywords: SARS-CoV-2 and COVID-19; comparative genetics; positive selection; primates and bats; virus–host coevolution.
Conflict of interest statement
The authors declare no competing interest.
Figures

Similar articles
-
Evolutionary Arms Race between Virus and Host Drives Genetic Diversity in Bat Severe Acute Respiratory Syndrome-Related Coronavirus Spike Genes.J Virol. 2020 Sep 29;94(20):e00902-20. doi: 10.1128/JVI.00902-20. Print 2020 Sep 29. J Virol. 2020. PMID: 32699095 Free PMC article.
-
Molecular evolution and phylogenetic analysis of SARS-CoV-2 and hosts ACE2 protein suggest Malayan pangolin as intermediary host.Braz J Microbiol. 2020 Dec;51(4):1593-1599. doi: 10.1007/s42770-020-00321-1. Epub 2020 Jun 26. Braz J Microbiol. 2020. PMID: 32592038 Free PMC article.
-
Comparative genomic analysis reveals varying levels of mammalian adaptation to coronavirus infections.PLoS Comput Biol. 2021 Nov 18;17(11):e1009560. doi: 10.1371/journal.pcbi.1009560. eCollection 2021 Nov. PLoS Comput Biol. 2021. PMID: 34793437 Free PMC article.
-
[Source of the COVID-19 pandemic: ecology and genetics of coronaviruses (Betacoronavirus: Coronaviridae) SARS-CoV, SARS-CoV-2 (subgenus Sarbecovirus), and MERS-CoV (subgenus Merbecovirus).].Vopr Virusol. 2020;65(2):62-70. doi: 10.36233/0507-4088-2020-65-2-62-70. Vopr Virusol. 2020. PMID: 32515561 Review. Russian.
-
Genetic variation analyses indicate conserved SARS-CoV-2-host interaction and varied genetic adaptation in immune response factors in modern human evolution.Dev Growth Differ. 2021 Apr;63(3):219-227. doi: 10.1111/dgd.12717. Epub 2021 Mar 21. Dev Growth Differ. 2021. PMID: 33595856 Free PMC article. Review.
Cited by
-
VANGL2 inhibits antiviral IFN-I signaling by targeting TBK1 for autophagic degradation.Sci Adv. 2023 Jun 23;9(25):eadg2339. doi: 10.1126/sciadv.adg2339. Epub 2023 Jun 23. Sci Adv. 2023. PMID: 37352355 Free PMC article.
-
Adaptive duplication and genetic diversification of protein kinase R contribute to the specificity of bat-virus interactions.Sci Adv. 2022 Nov 25;8(47):eadd7540. doi: 10.1126/sciadv.add7540. Epub 2022 Nov 23. Sci Adv. 2022. PMID: 36417524 Free PMC article.
-
Evolutionary and functional analyses reveal a role for the RHIM in tuning RIPK3 activity across vertebrates.Elife. 2025 May 28;13:RP102301. doi: 10.7554/eLife.102301. Elife. 2025. PMID: 40434815 Free PMC article.
-
ACE2-independent sarbecovirus cell entry can be supported by TMPRSS2-related enzymes and can reduce sensitivity to antibody-mediated neutralization.PLoS Pathog. 2024 Nov 13;20(11):e1012653. doi: 10.1371/journal.ppat.1012653. eCollection 2024 Nov. PLoS Pathog. 2024. PMID: 39536058 Free PMC article.
-
Evolutionary and functional analyses reveal a role for the RHIM in tuning RIPK3 activity across vertebrates.bioRxiv [Preprint]. 2025 Jan 31:2024.05.09.593370. doi: 10.1101/2024.05.09.593370. bioRxiv. 2025. Update in: Elife. 2025 May 28;13:RP102301. doi: 10.7554/eLife.102301. PMID: 39149247 Free PMC article. Updated. Preprint.
References
-
- Temmam S., et al. , Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 604, 330–336 (2022). - PubMed
-
- Christie M. J., et al. , Of bats and men: Immunomodulatory treatment options for COVID-19 guided by the immunopathology of SARS-CoV-2 infection. Sci. Immunol. 6, eabd0205 (2021). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
