

Combined MEK and STAT3 Inhibition Uncovers Stromal Plasticity by Enriching for Cancer-Associated Fibroblasts With Mesenchymal Stem Cell-Like Features to Overcome Immunotherapy Resistance in Pancreatic Cancer
- PMID: 35948109
- PMCID: PMC10257389
- DOI: 10.1053/j.gastro.2022.07.076
Combined MEK and STAT3 Inhibition Uncovers Stromal Plasticity by Enriching for Cancer-Associated Fibroblasts With Mesenchymal Stem Cell-Like Features to Overcome Immunotherapy Resistance in Pancreatic Cancer
Abstract
Background & aims: We have shown that reciprocally activated rat sarcoma (RAS)/mitogen-activated protein kinase/extracellular signal-regulated kinase (MEK) and Janus kinase/signal transducer and activator of transcription 3 (STAT3) pathways mediate therapeutic resistance in pancreatic ductal adenocarcinoma (PDAC), while combined MEK and STAT3 inhibition (MEKi+STAT3i) overcomes such resistance and alters stromal architecture. We now determine whether MEKi+STAT3i reprograms the cancer-associated fibroblast (CAF) and immune microenvironment to overcome resistance to immune checkpoint inhibition in PDAC.
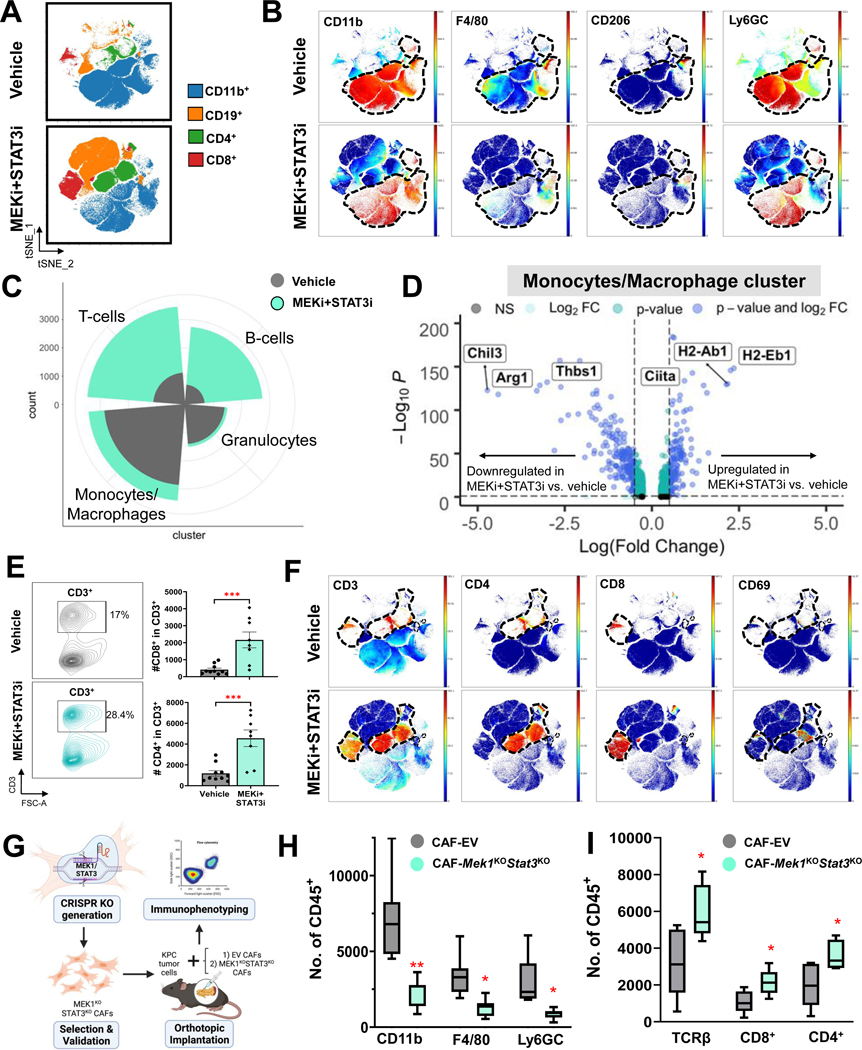
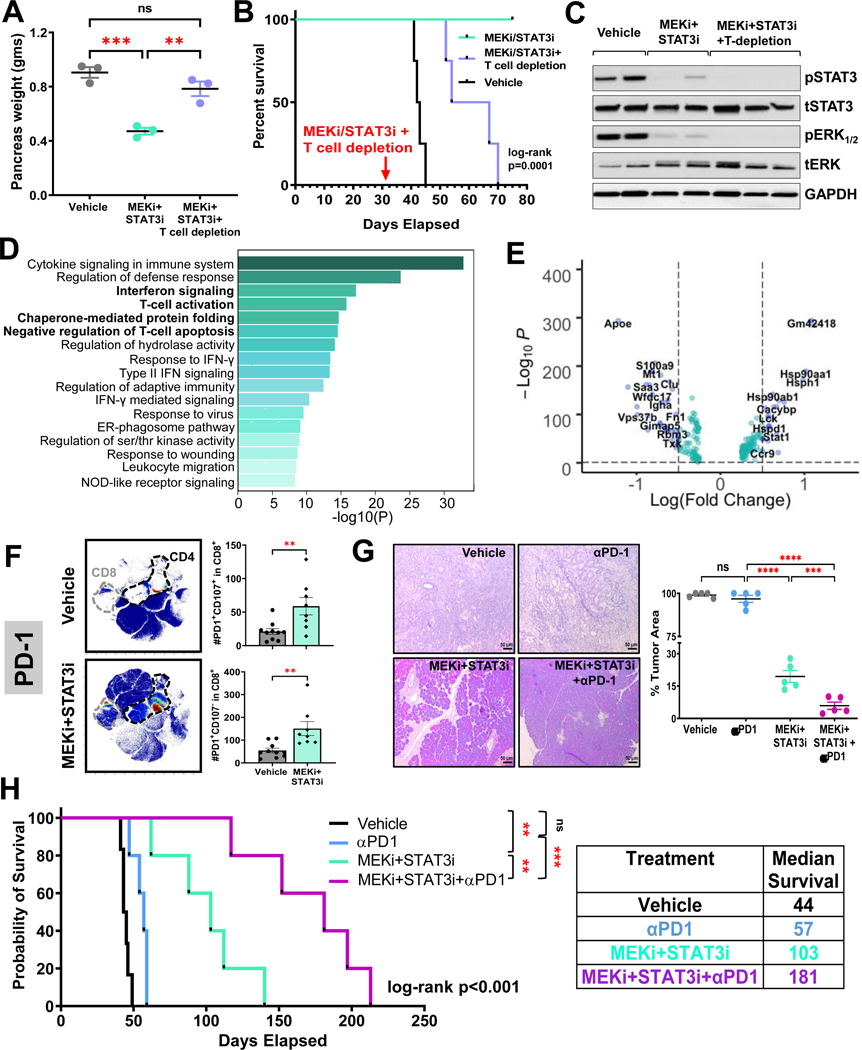
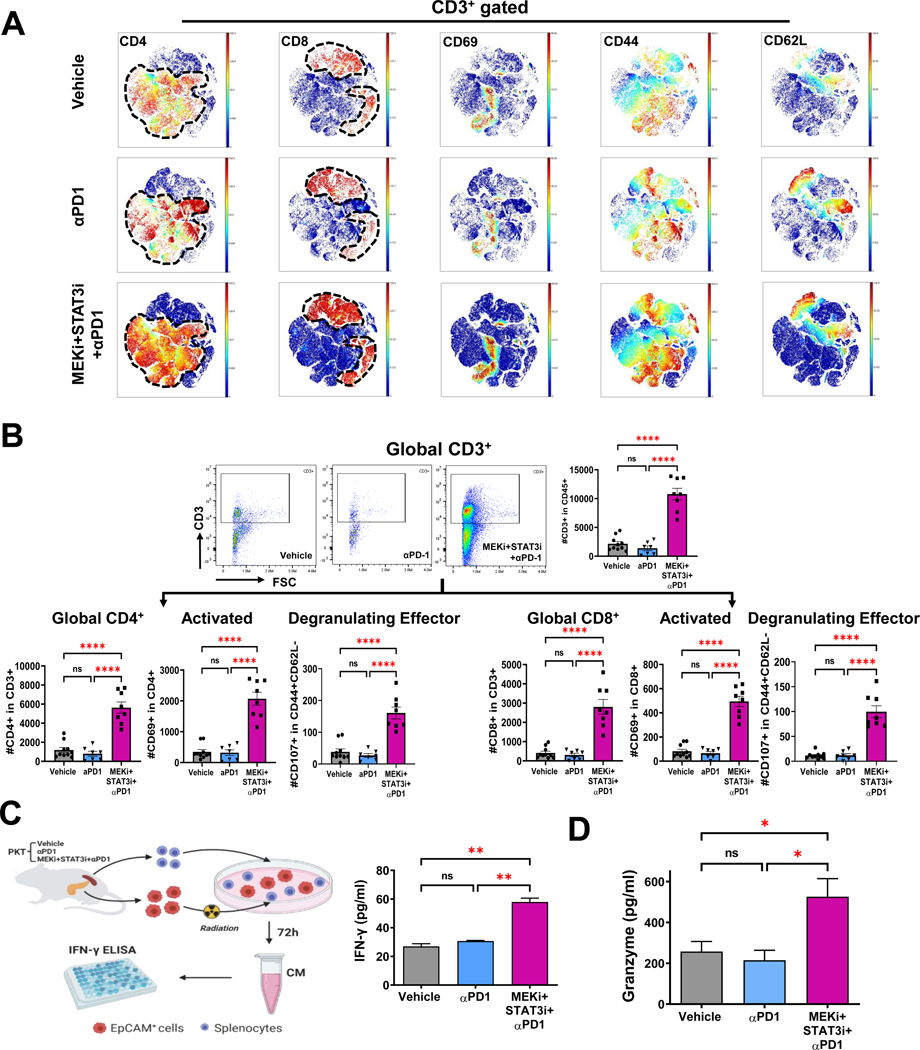
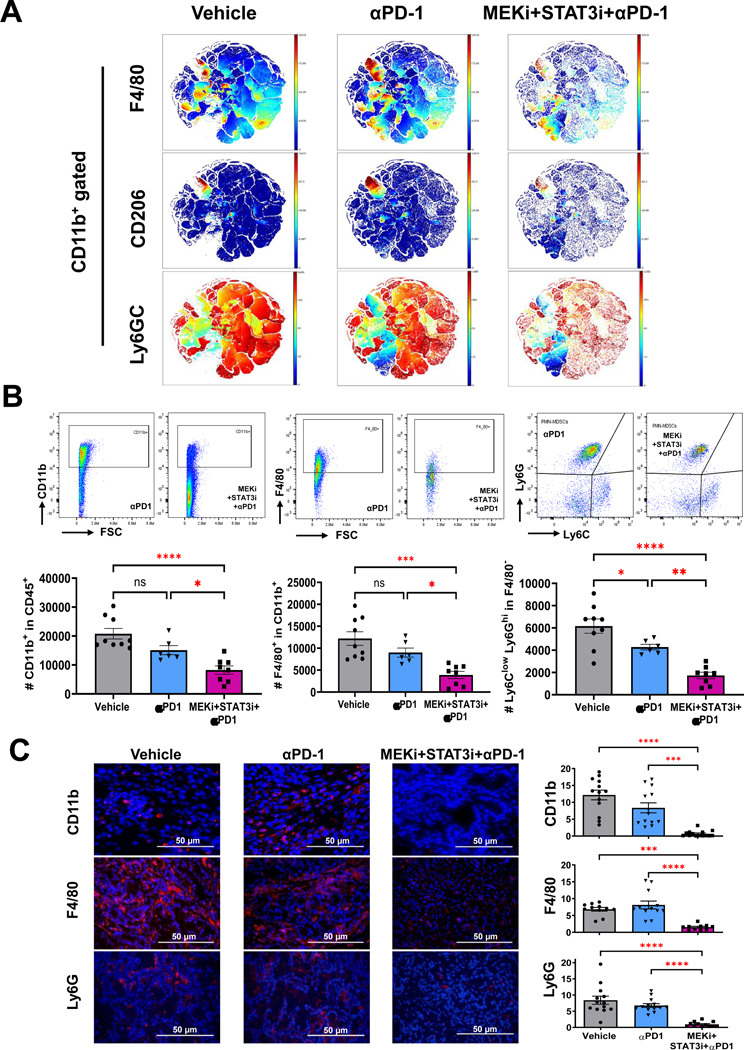
Methods: CAF and immune cell transcriptomes in MEKi (trametinib)+STAT3i (ruxolitinib)-treated vs vehicle-treated Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) tumors were examined via single-cell RNA sequencing (scRNAseq). Clustered regularly interspaced short palindromic repeats/clustered regularly interspaced short palindromic repeats associated protein 9 silencing of CAF-restricted Map2k1/Mek1 or Stat3, or both, enabled interrogation of CAF-dependent effects on immunologic remodeling in orthotopic models. Tumor growth, survival, and immune profiling via mass cytometry by time-of-flight were examined in PKT mice treated with vehicle, anti-programmed cell death protein 1 (PD-1) monotherapy, and MEKi+STAT3i combined with anti-PD1.
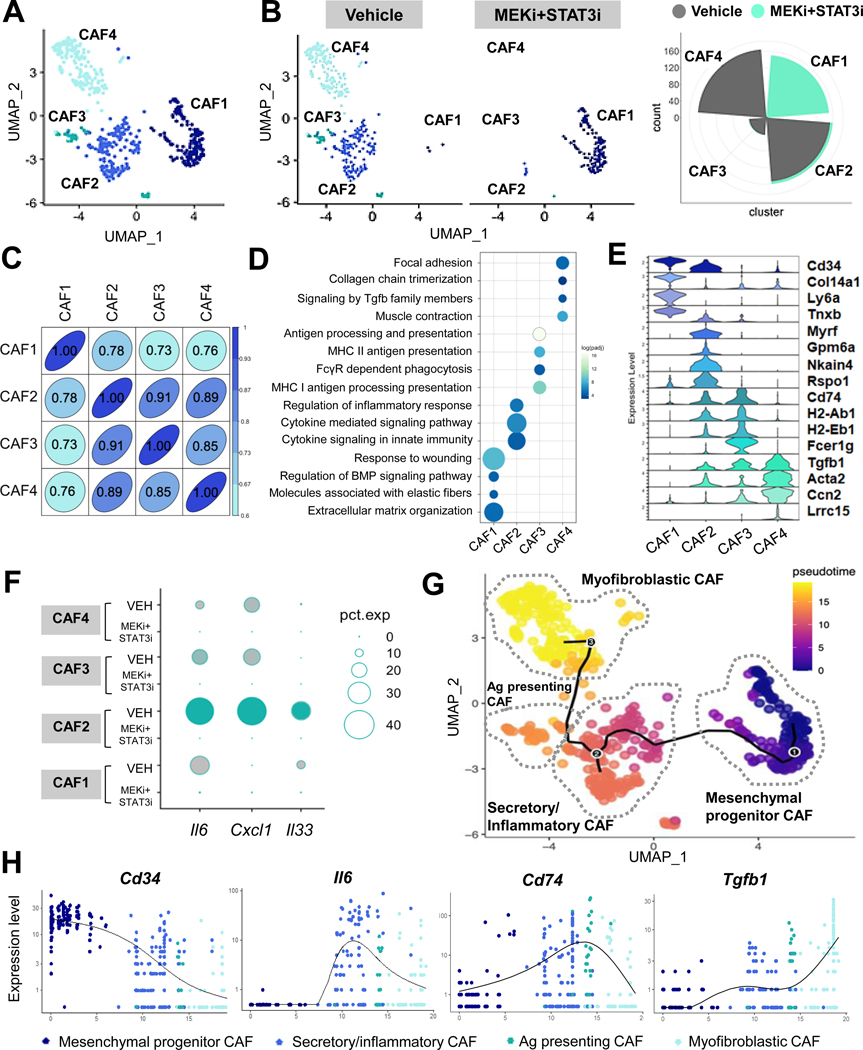
Results: MEKi+STAT3i attenuates Il6/Cxcl1-expressing proinflammatory and Lrrc15-expressing myofibroblastic CAF phenotypes while enriching for Ly6a/Cd34-expressing CAFs exhibiting mesenchymal stem cell-like features via scRNAseq in PKT mice. This CAF plasticity is associated with M2-to-M1 reprogramming of tumor-associated macrophages, and enhanced trafficking of cluster of differentiation 8+ T cells, which exhibit distinct effector transcriptional programs. These MEKi+STAT3i-induced effects appear CAF-dependent, because CAF-restricted Mek1/Stat3 silencing mitigates inflammatory-CAF polarization and myeloid infiltration in vivo. Addition of MEKi+STAT3i to PD-1 blockade not only dramatically improves antitumor responses and survival in PKT mice but also augments recruitment of activated/memory T cells while improving their degranulating and cytotoxic capacity compared with anti-PD-1 monotherapy. Importantly, treatment of a patient who has chemotherapy-refractory metastatic PDAC with MEKi (trametinib), STAT3i (ruxolitinib), and PD-1 inhibitor (nivolumab) yielded clinical benefit.
Conclusions: Combined MEKi+STAT3i mitigates stromal inflammation and enriches for CAF phenotypes with mesenchymal stem cell-like properties to overcome immunotherapy resistance in PDAC.
Keywords: Cancer-Associated Fibroblast; Heterogeneity; Immune Checkpoint Inhibition; Immunotherapy; MEK; Mesenchymal Stem Cell; Pancreatic Ductal Adenocarcinoma; STAT3; Stromal Plasticity.
Copyright © 2022 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures


References
-
- Beatty GL, Eghbali S, Kim R. Deploying Immunotherapy in Pancreatic Cancer: Defining Mechanisms of Response and Resistance. Am Soc Clin Oncol Educ Book 2017;37:267–278. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
