

Single-cell multi-omics of human preimplantation embryos shows susceptibility to glucocorticoids
- PMID: 35948369
- PMCID: PMC9528977
- DOI: 10.1101/gr.276665.122
Single-cell multi-omics of human preimplantation embryos shows susceptibility to glucocorticoids
Abstract
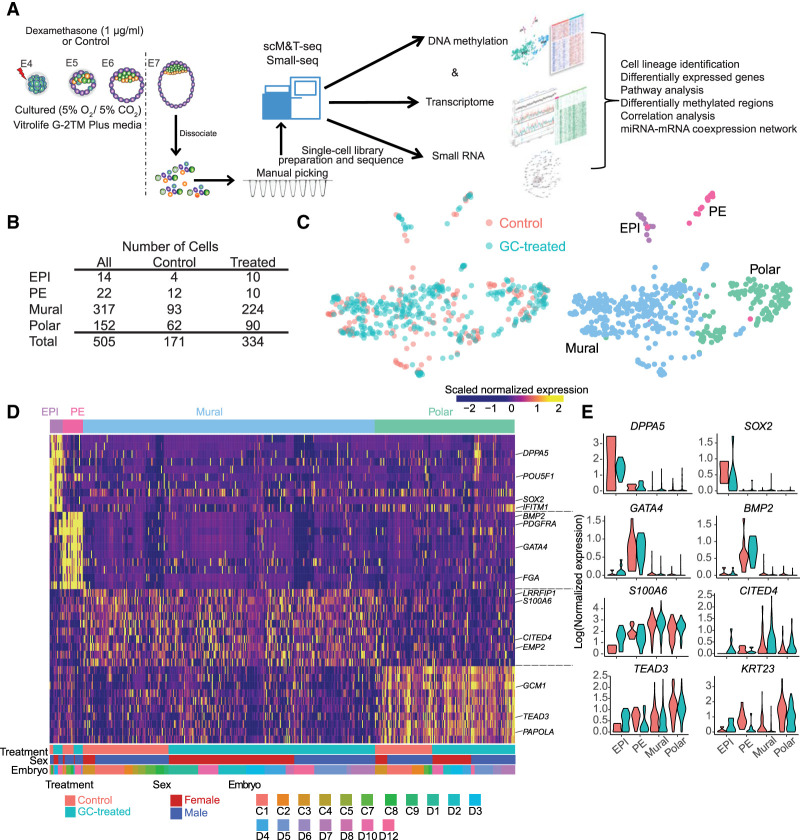
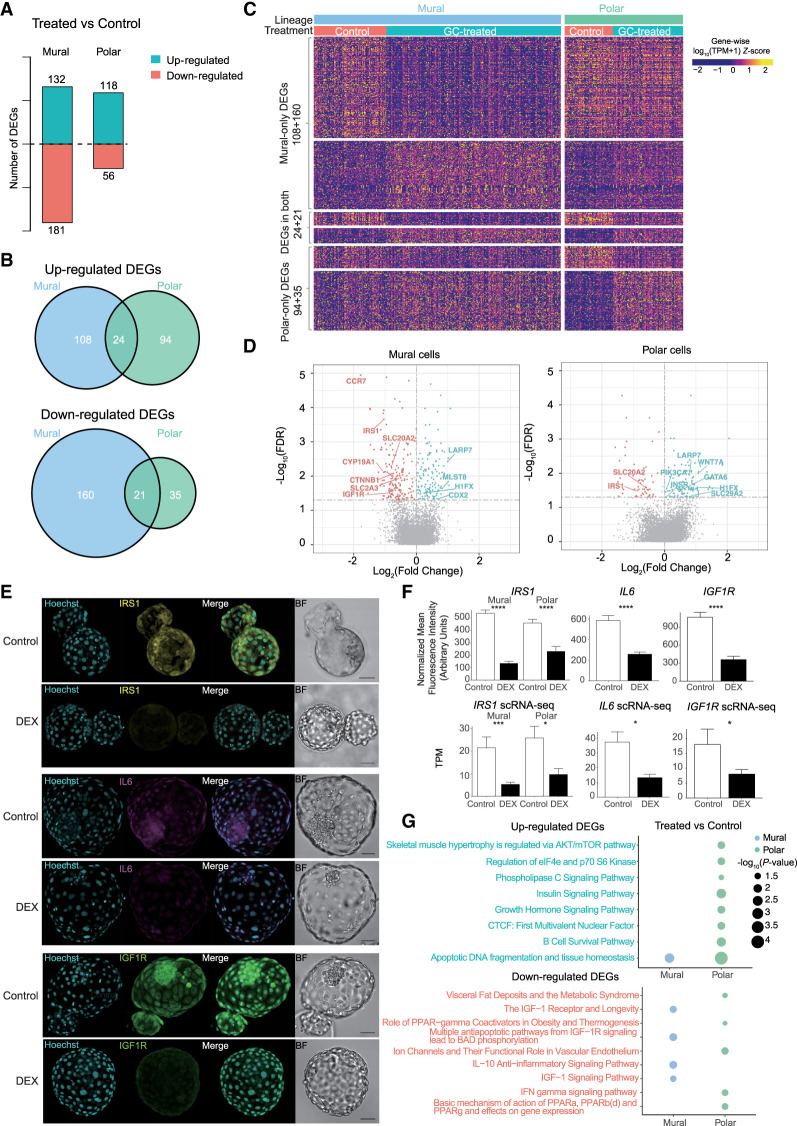
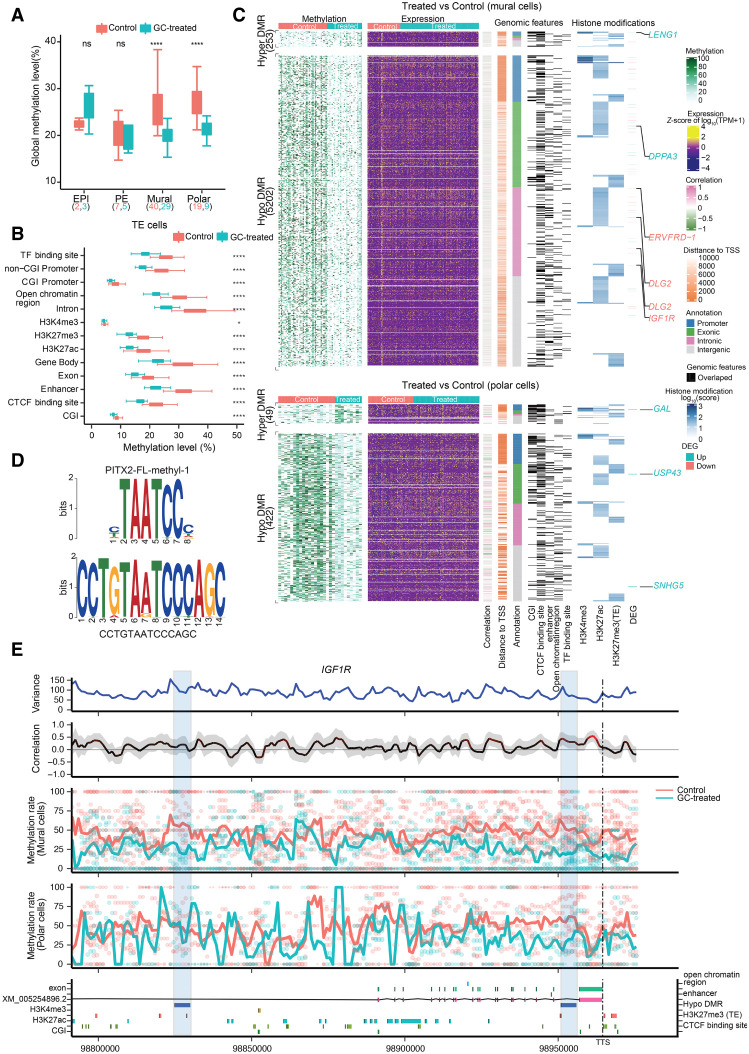
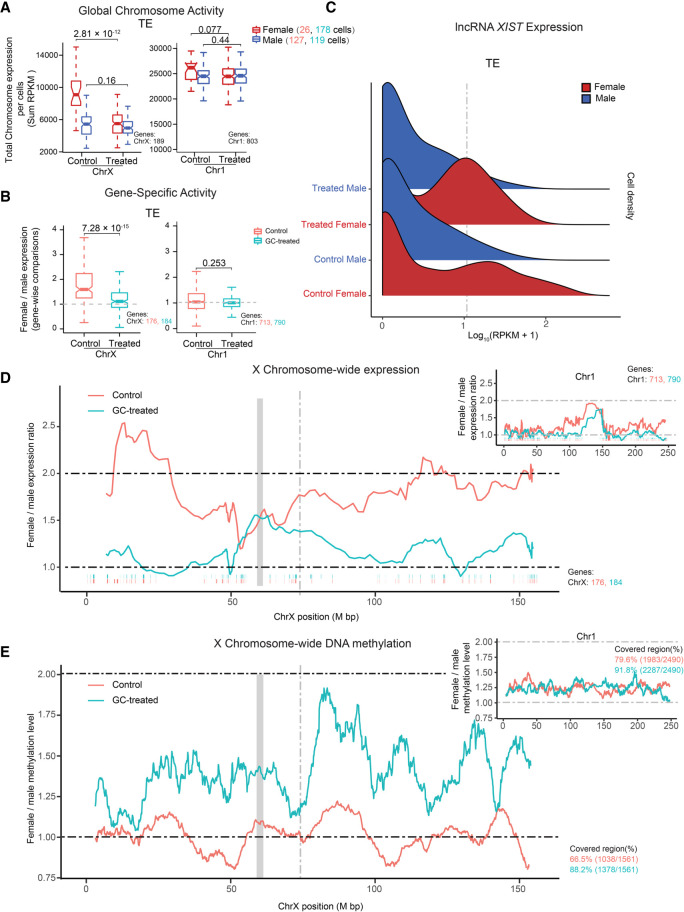
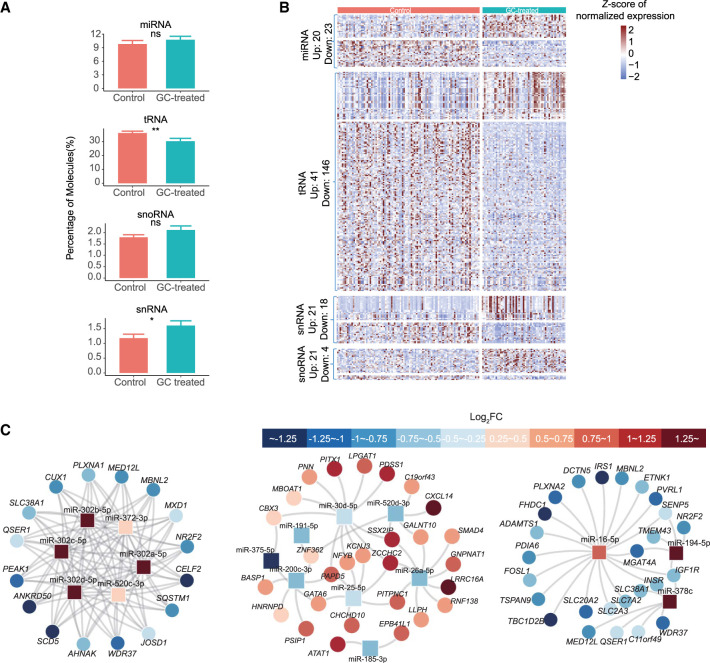
The preconceptual, intrauterine, and early life environments can have a profound and long-lasting impact on the developmental trajectories and health outcomes of the offspring. Given the relatively low success rates of assisted reproductive technologies (ART; ∼25%), additives and adjuvants, such as glucocorticoids, are used to improve the success rate. Considering the dynamic developmental events that occur during this window, these exposures may alter blastocyst formation at a molecular level, and as such, affect not only the viability of the embryo and the ability of the blastocyst to implant, but also the developmental trajectory of the first three cell lineages, ultimately influencing the physiology of the embryo. In this study, we present a comprehensive single-cell transcriptome, methylome, and small RNA atlas in the day 7 human embryo. We show that, despite no change in morphology and developmental features, preimplantation glucocorticoid exposure reprograms the molecular profile of the trophectoderm (TE) lineage, and these changes are associated with an altered metabolic and inflammatory response. Our data also suggest that glucocorticoids can precociously mature the TE sublineages, supported by the presence of extravillous trophoblast markers in the polar sublineage and presence of X Chromosome dosage compensation. Further, we have elucidated that epigenetic regulation-DNA methylation and microRNAs (miRNAs)-likely underlies the transcriptional changes observed. This study suggests that exposures to exogenous compounds during preimplantation may unintentionally reprogram the human embryo, possibly leading to suboptimal development and longer-term health outcomes.
© 2022 Zhao et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical