

Pyramidal neuron subtype diversity governs microglia states in the neocortex
- PMID: 35948630
- PMCID: PMC10502800
- DOI: 10.1038/s41586-022-05056-7
Pyramidal neuron subtype diversity governs microglia states in the neocortex
Abstract
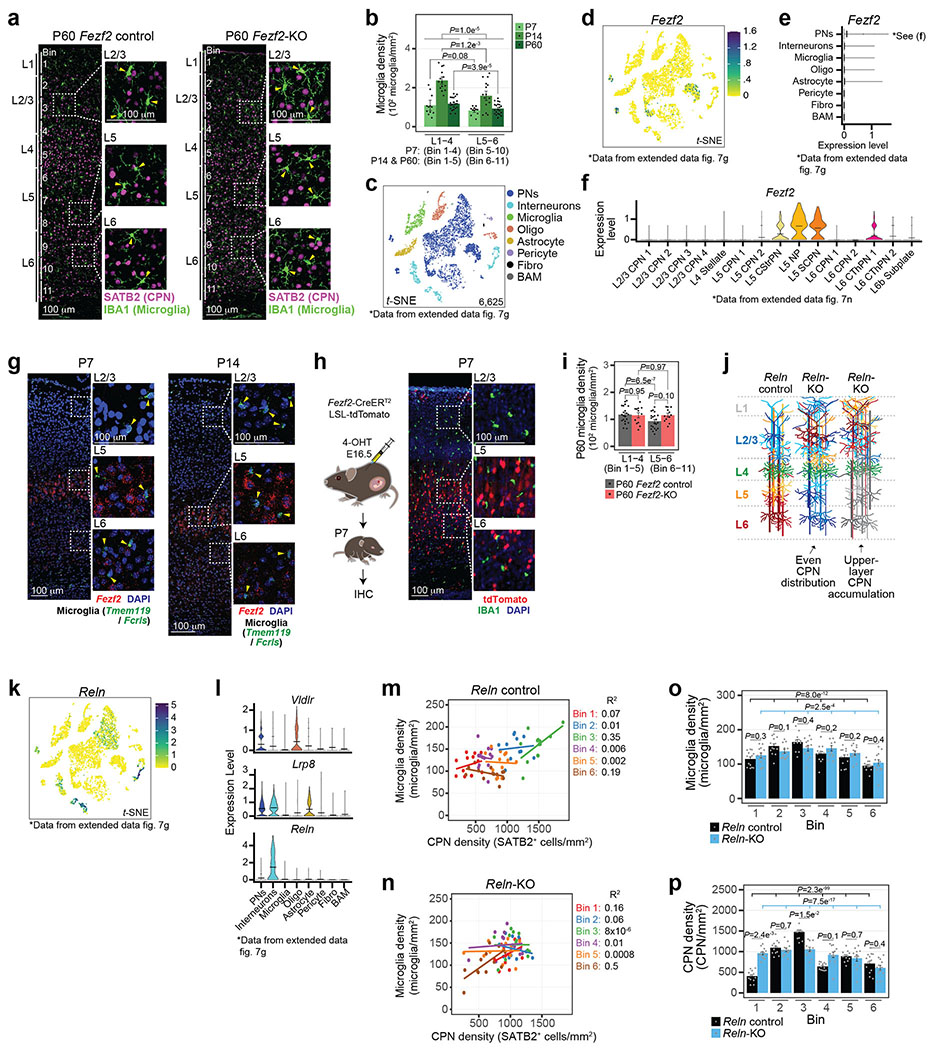
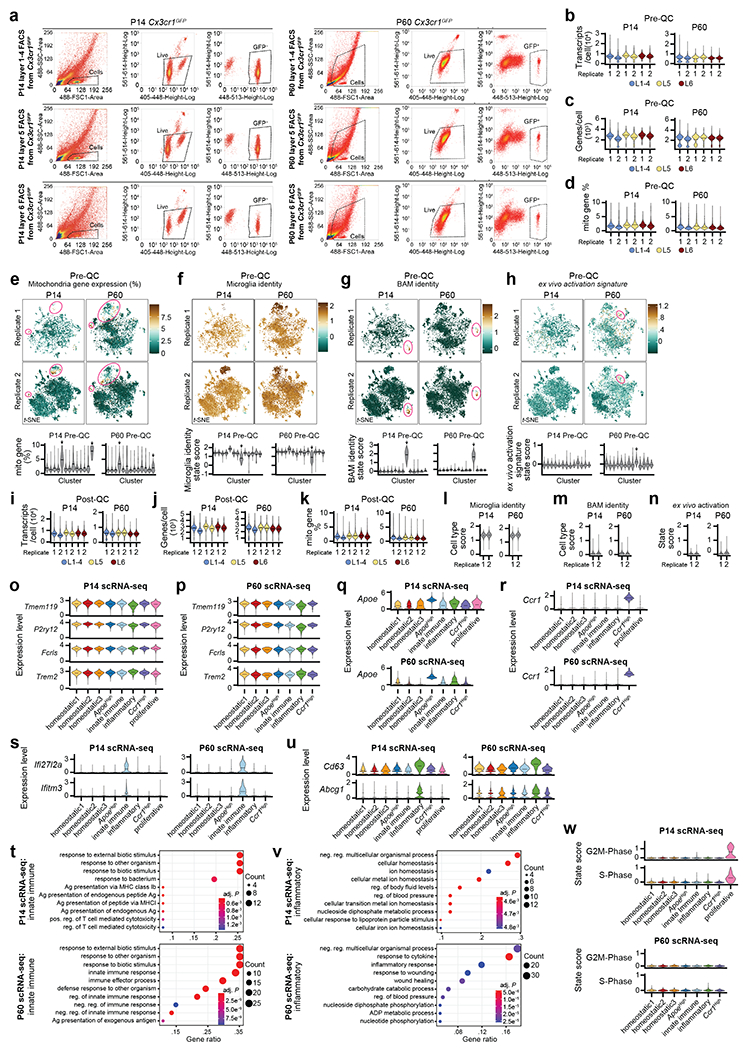
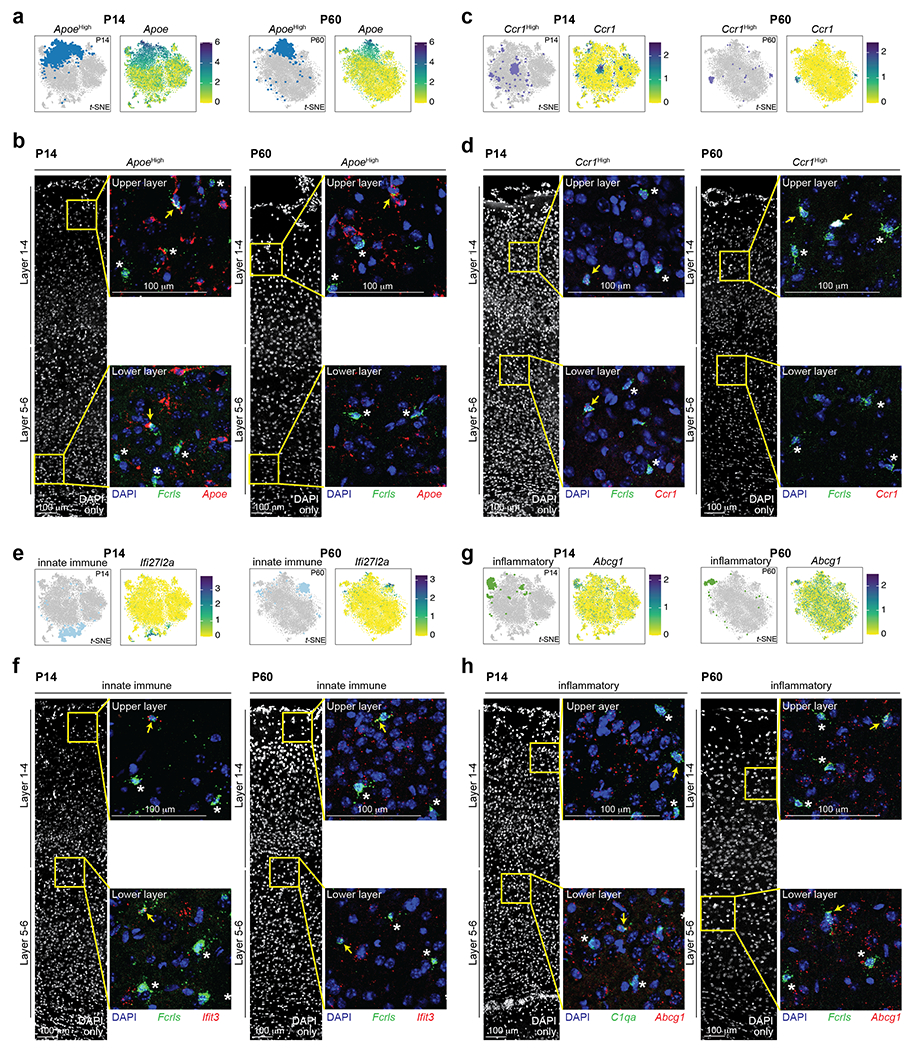
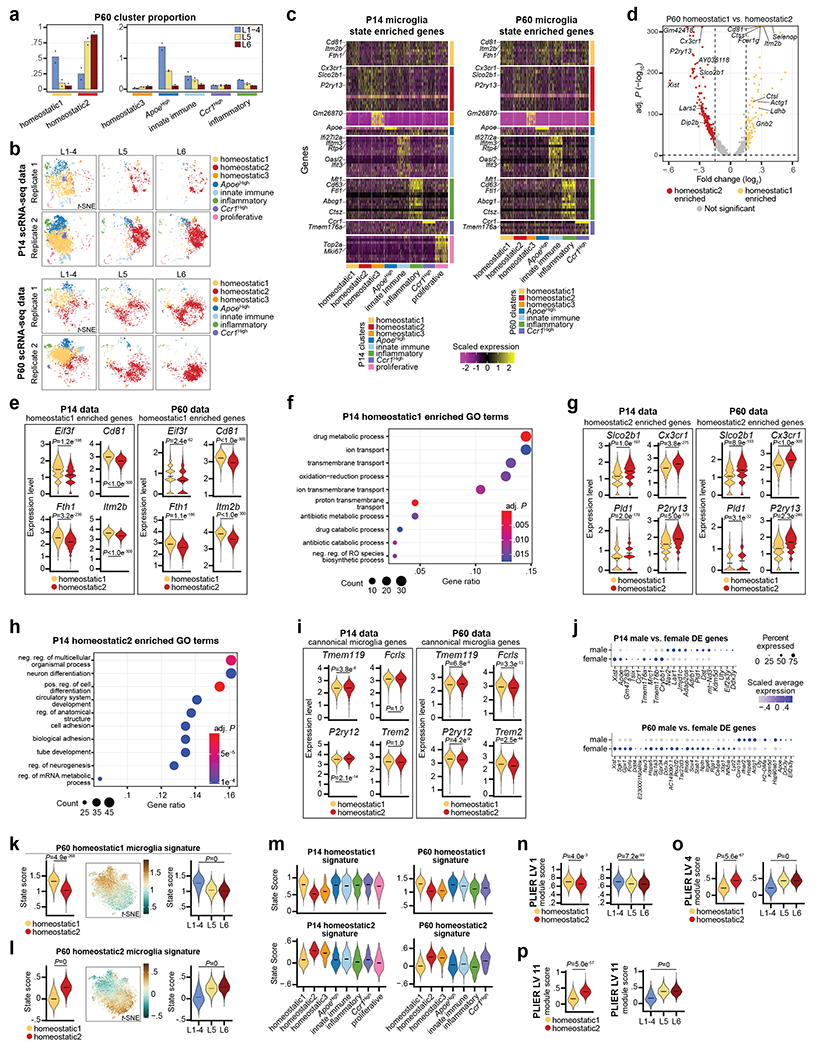
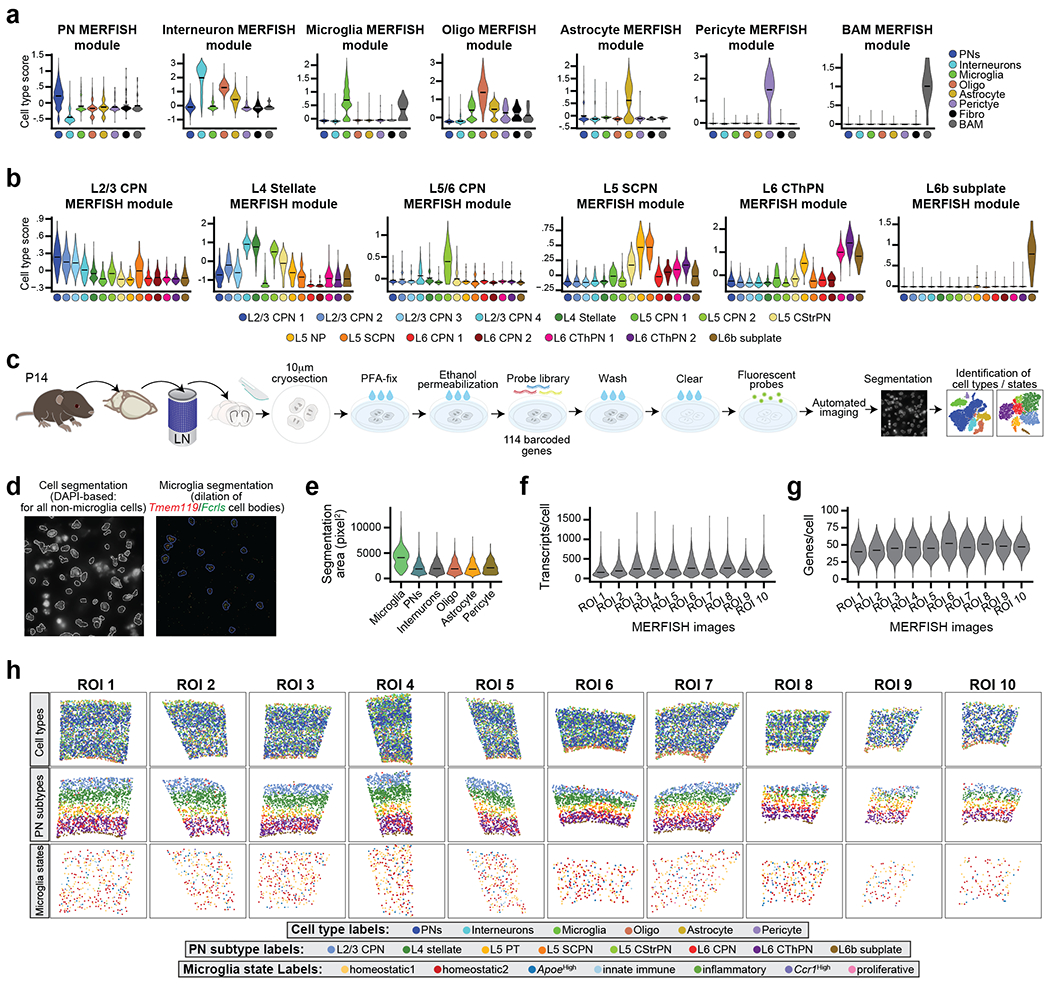
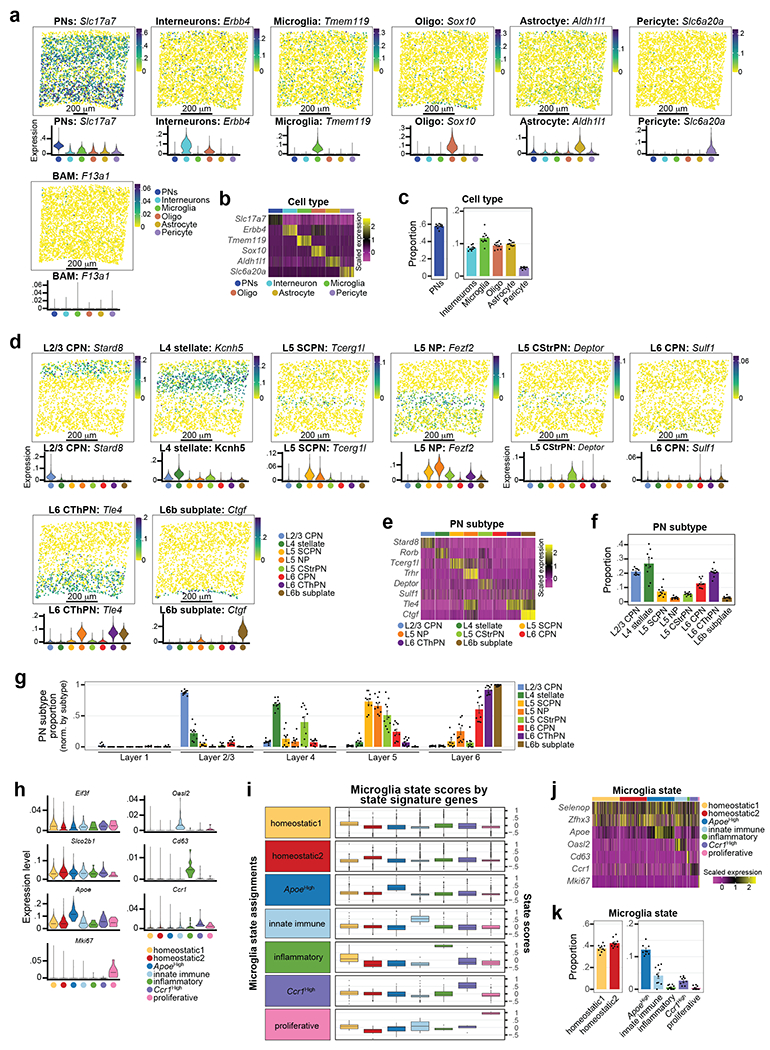
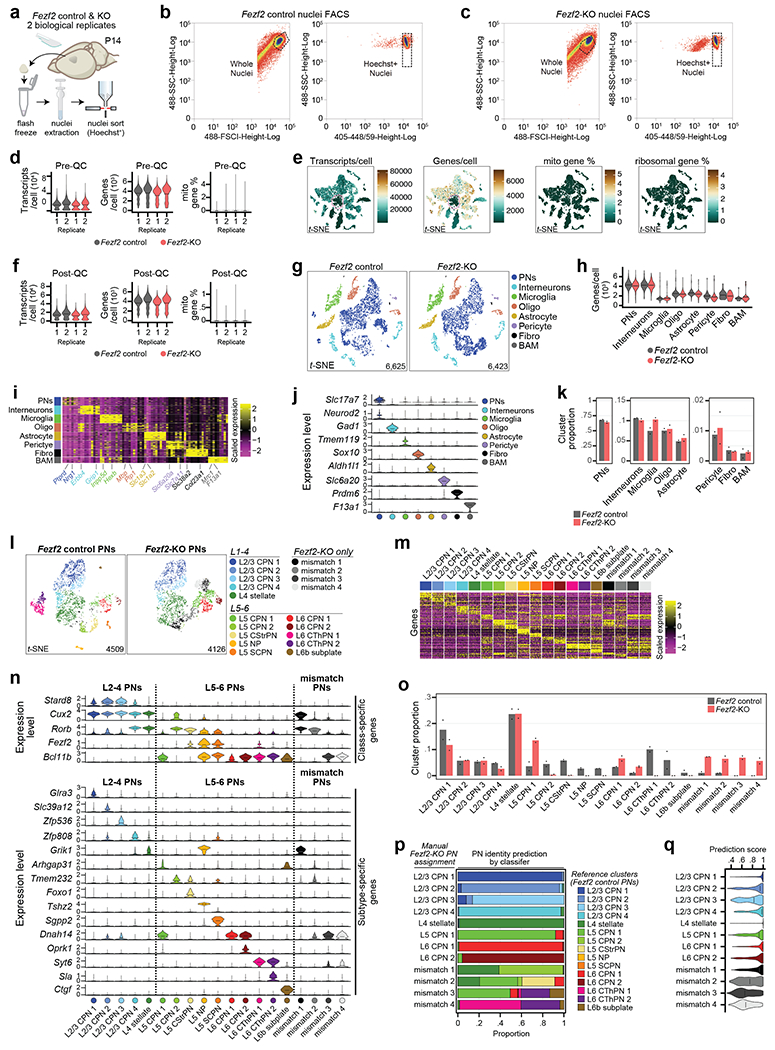
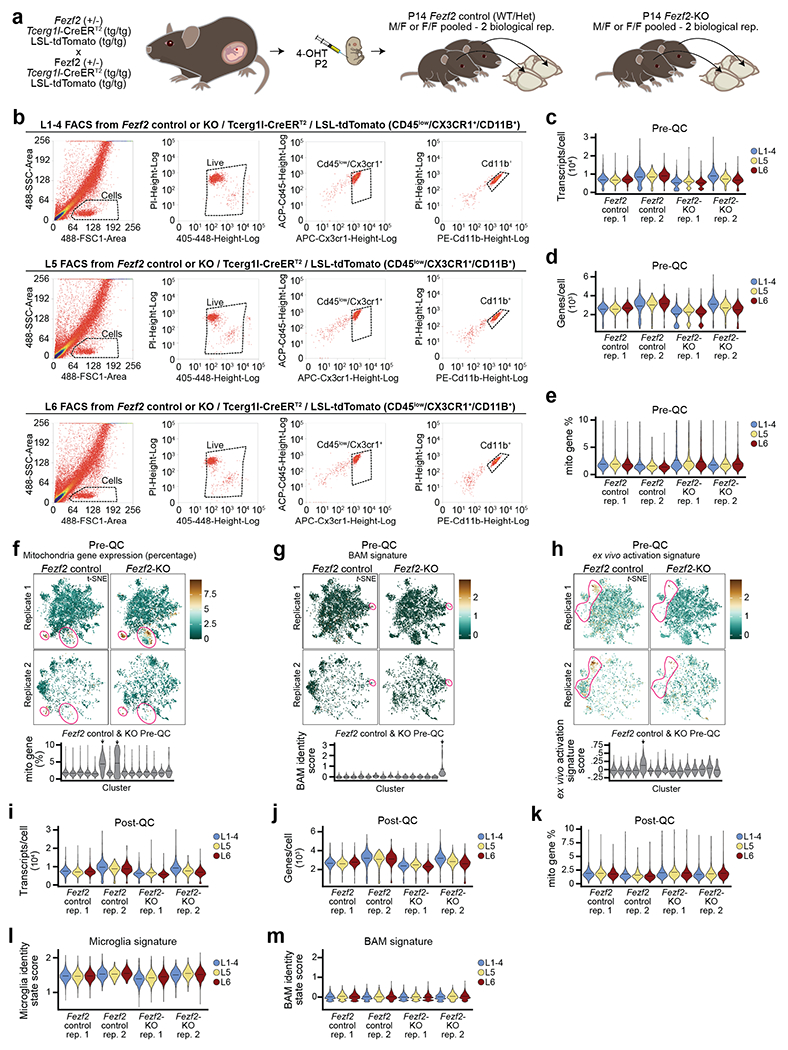
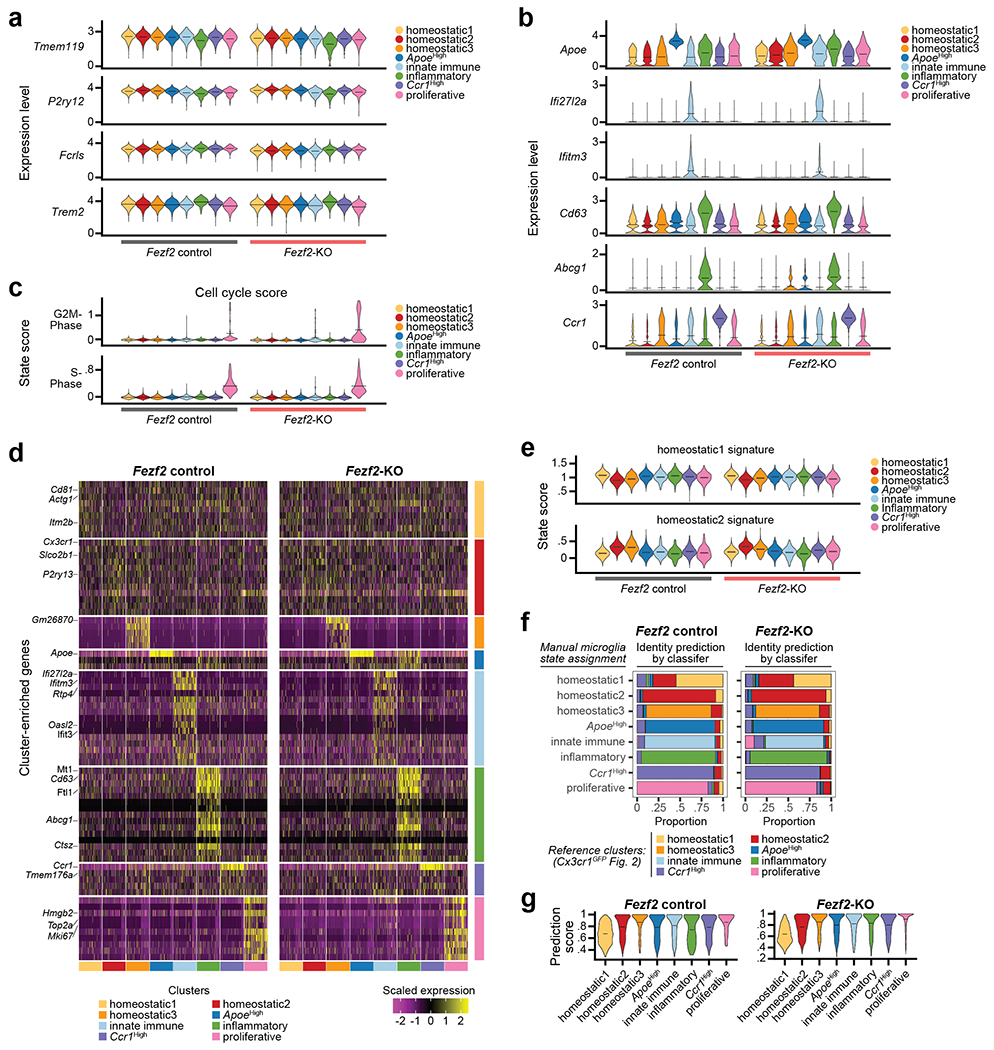
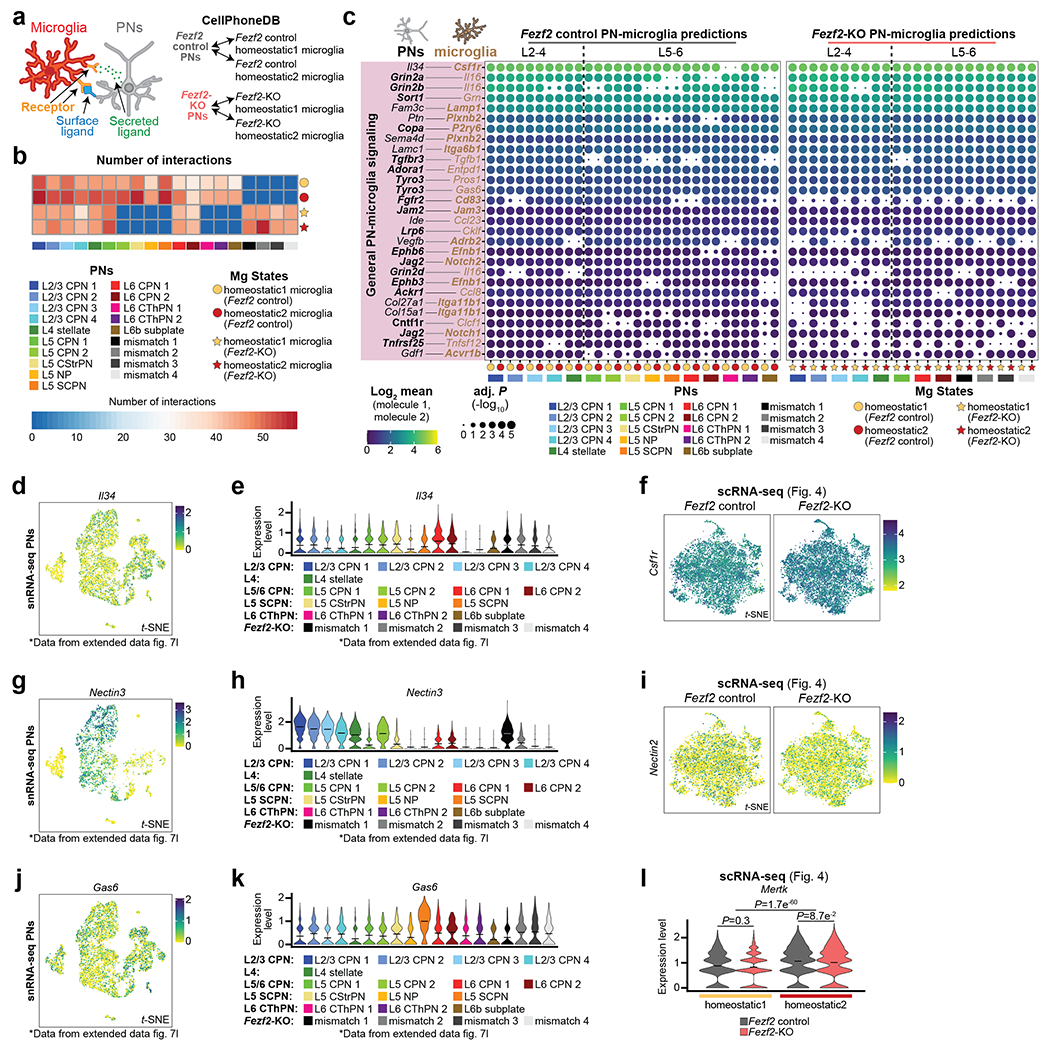
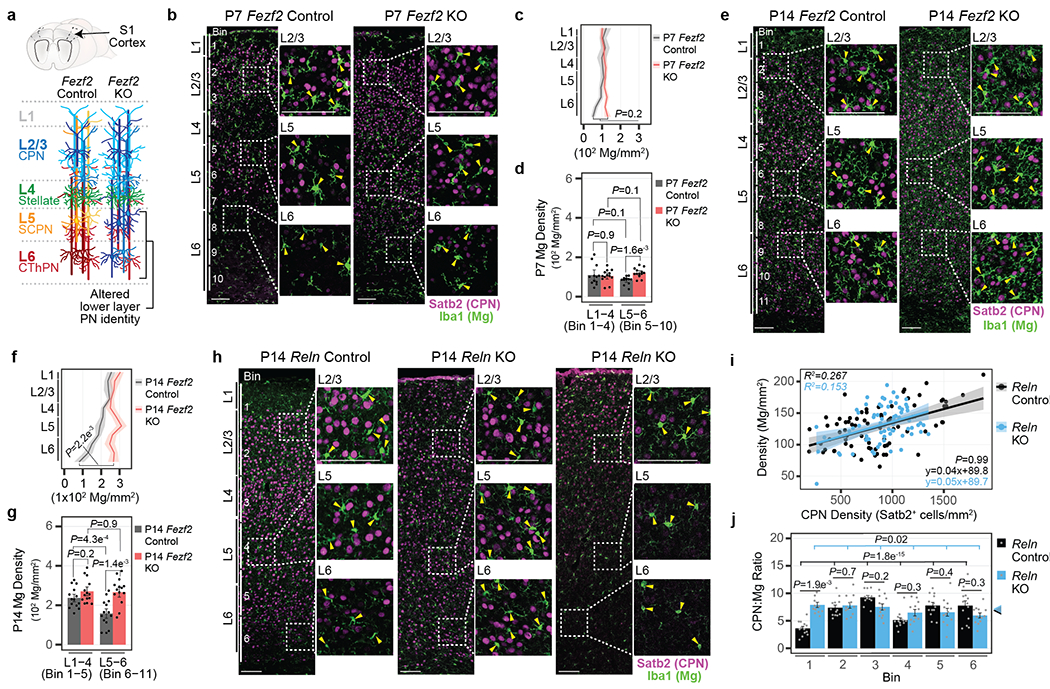
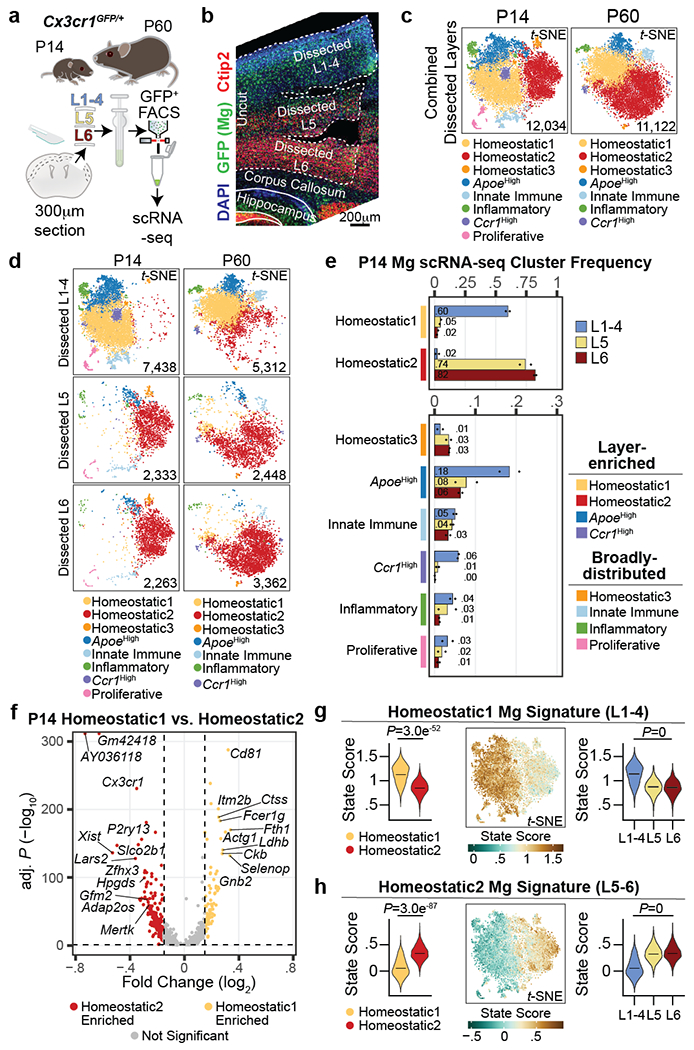
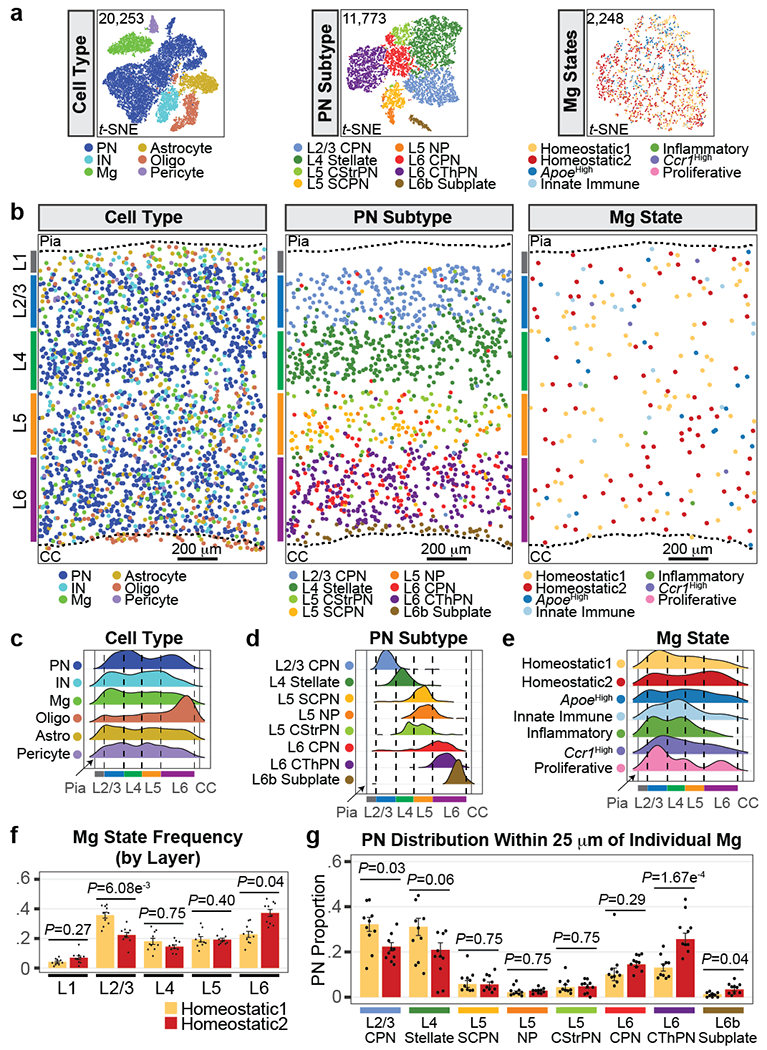
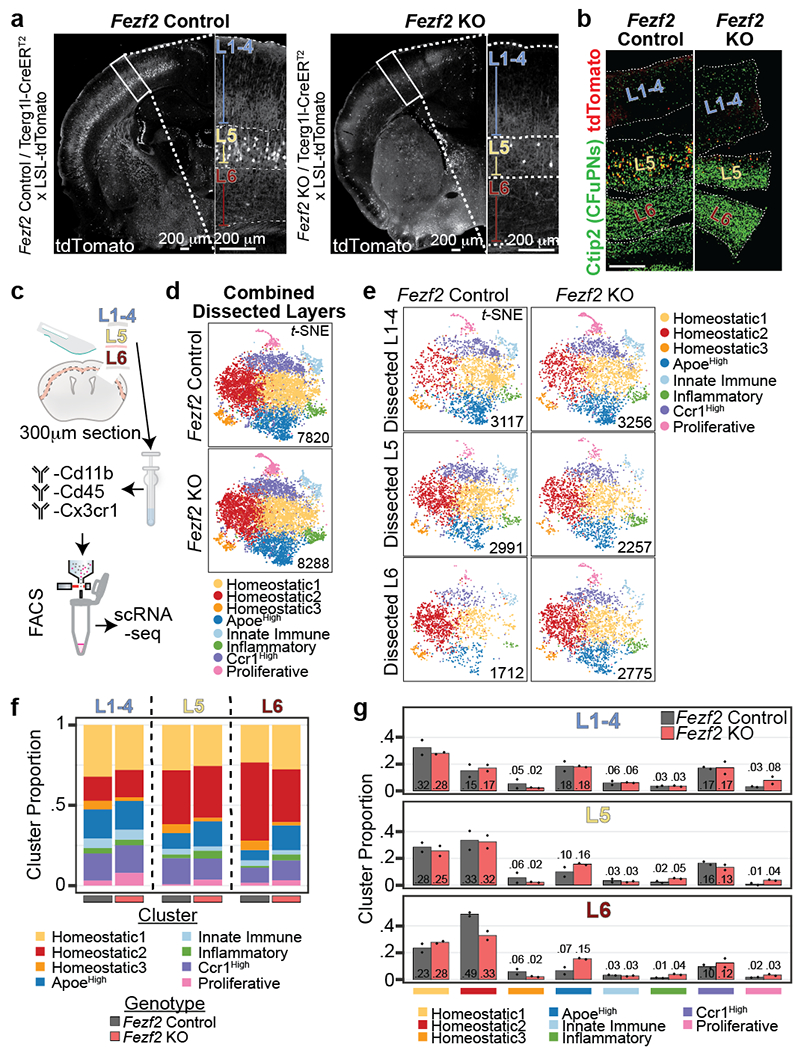
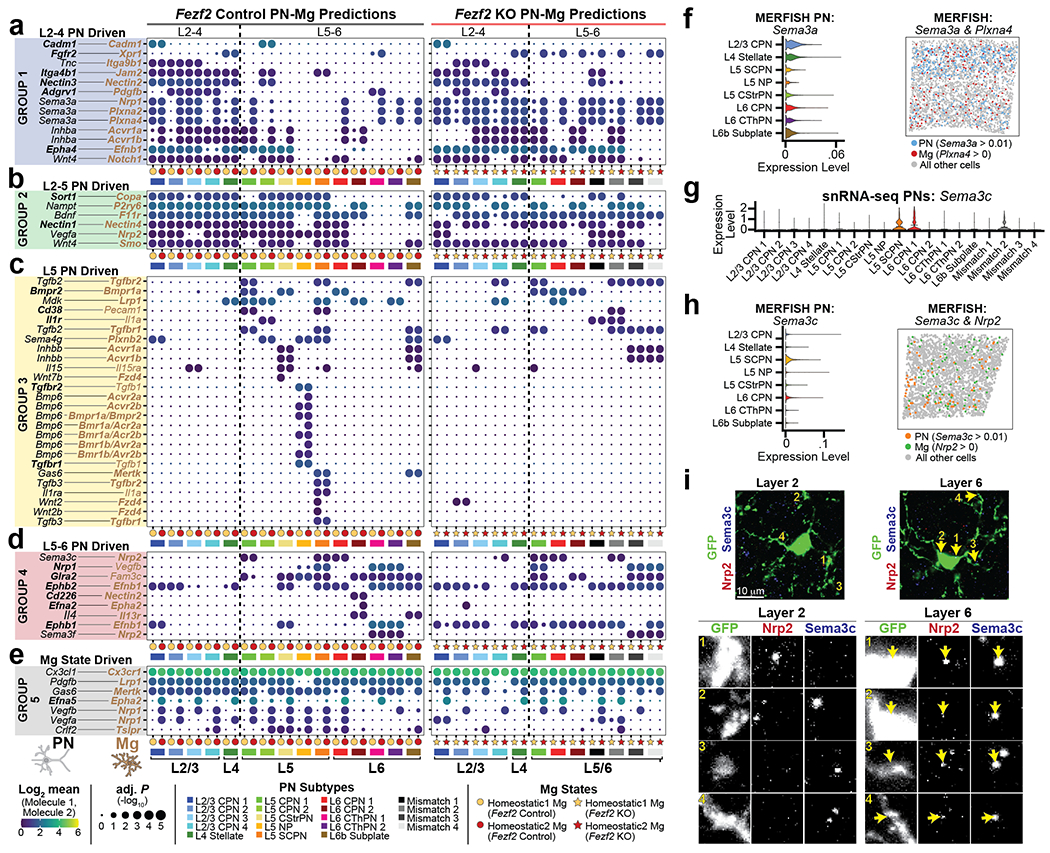
Microglia are specialized macrophages in the brain parenchyma that exist in multiple transcriptional states and reside within a wide range of neuronal environments1-4. However, how and where these states are generated remains poorly understood. Here, using the mouse somatosensory cortex, we demonstrate that microglia density and molecular state acquisition are determined by the local composition of pyramidal neuron classes. Using single-cell and spatial transcriptomic profiling, we unveil the molecular signatures and spatial distributions of diverse microglia populations and show that certain states are enriched in specific cortical layers, whereas others are broadly distributed throughout the cortex. Notably, conversion of deep-layer pyramidal neurons to an alternate class identity reconfigures the distribution of local, layer-enriched homeostatic microglia to match the new neuronal niche. Leveraging the transcriptional diversity of pyramidal neurons in the neocortex, we construct a ligand-receptor atlas describing interactions between individual pyramidal neuron subtypes and microglia states, revealing rules of neuron-microglia communication. Our findings uncover a fundamental role for neuronal diversity in instructing the acquisition of microglia states as a potential mechanism for fine-tuning neuroimmune interactions within the cortical local circuitry.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
COMPETING INTEREST
P.A. is a SAB member in System 1 Biosciences and Foresite Labs and is a co-founder of FL60. J.A.S. is an employee of Sana Biotechnology, Inc. as of 09/2022.
Figures
Comment in
-
Neuronal neighbours tune microglial identity.Nat Rev Neurosci. 2022 Oct;23(10):582-583. doi: 10.1038/s41583-022-00632-2. Nat Rev Neurosci. 2022. PMID: 36042345 No abstract available.
References
-
- Lawson LJ, Perry VH, Dri P & Gordon S Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39, 151–170 (1990). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
