

Diverse mutational landscapes in human lymphocytes
- PMID: 35948631
- PMCID: PMC9402440
- DOI: 10.1038/s41586-022-05072-7
Diverse mutational landscapes in human lymphocytes
Abstract
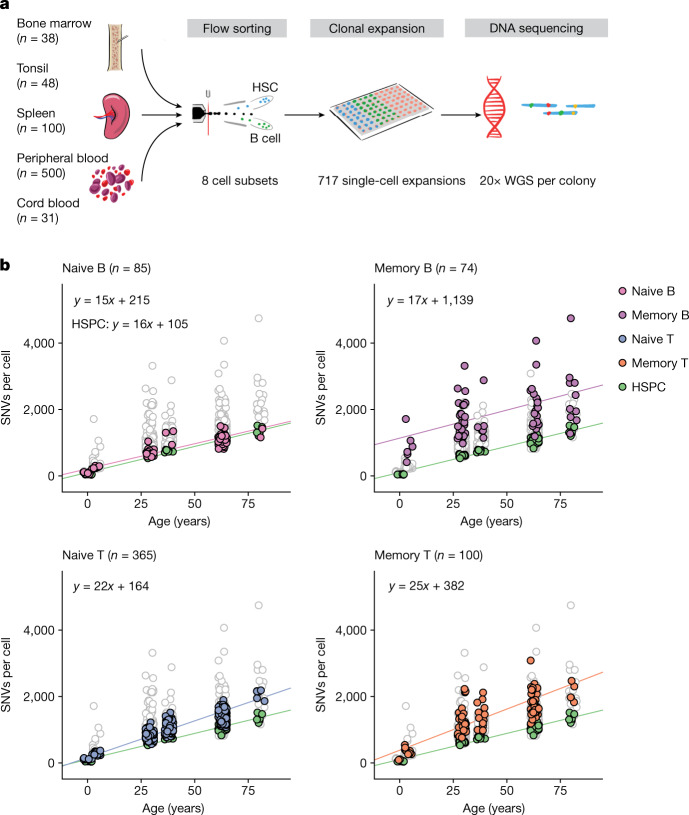
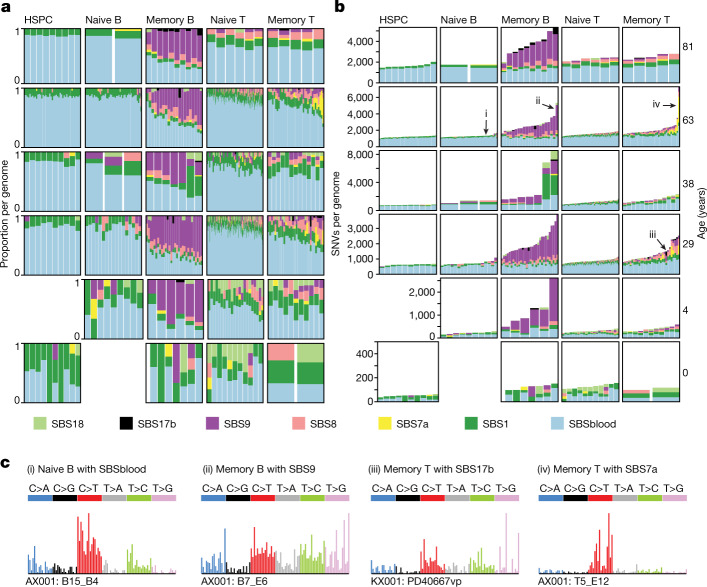
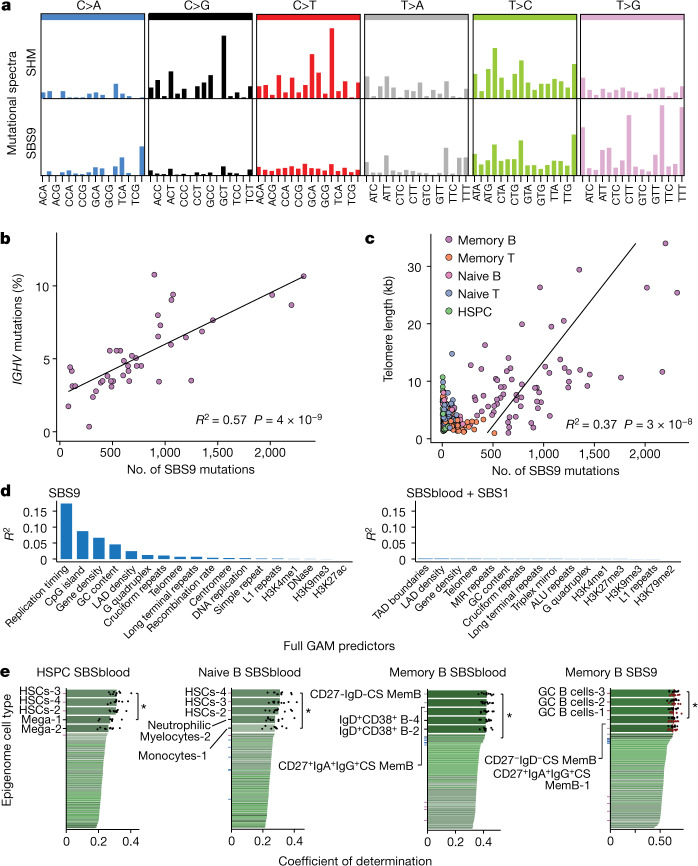
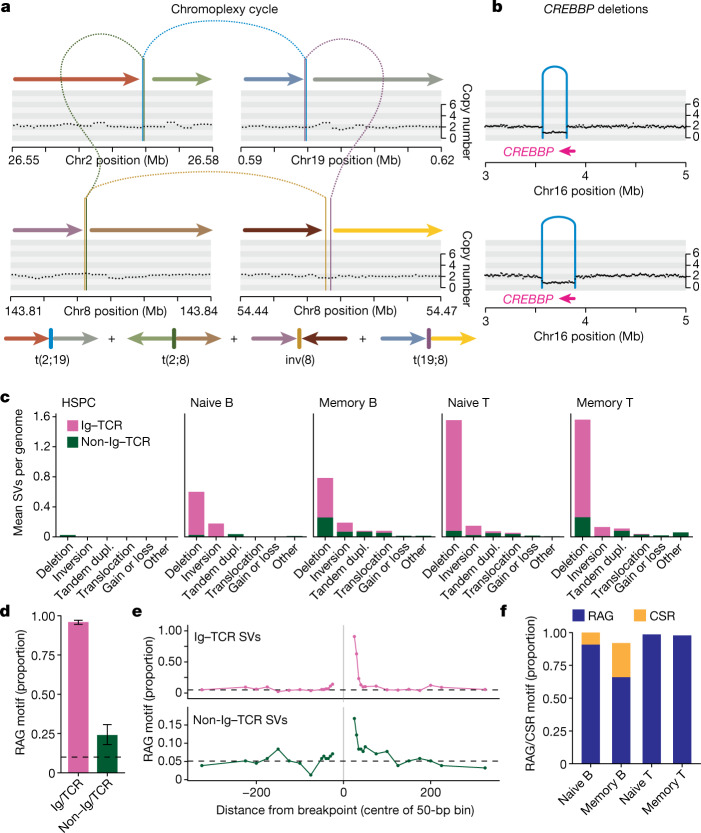
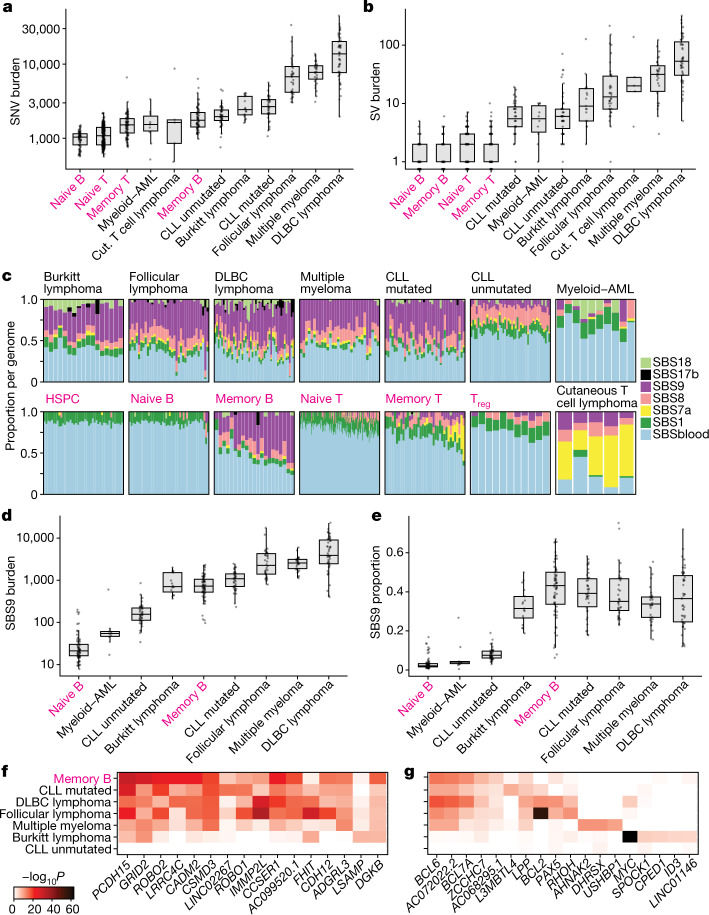
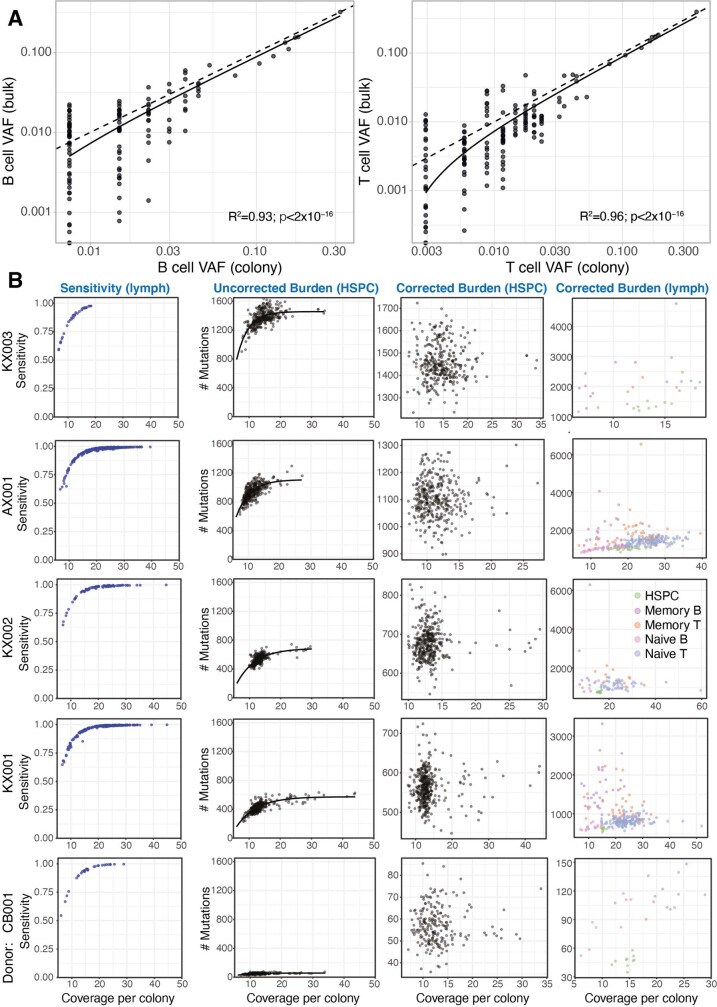
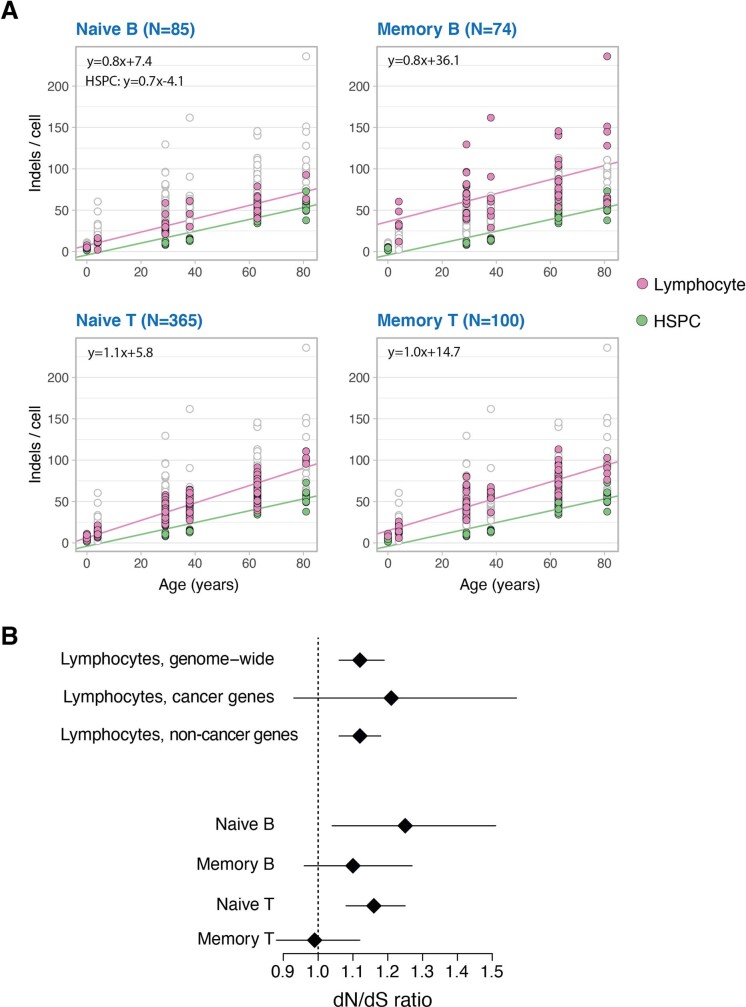
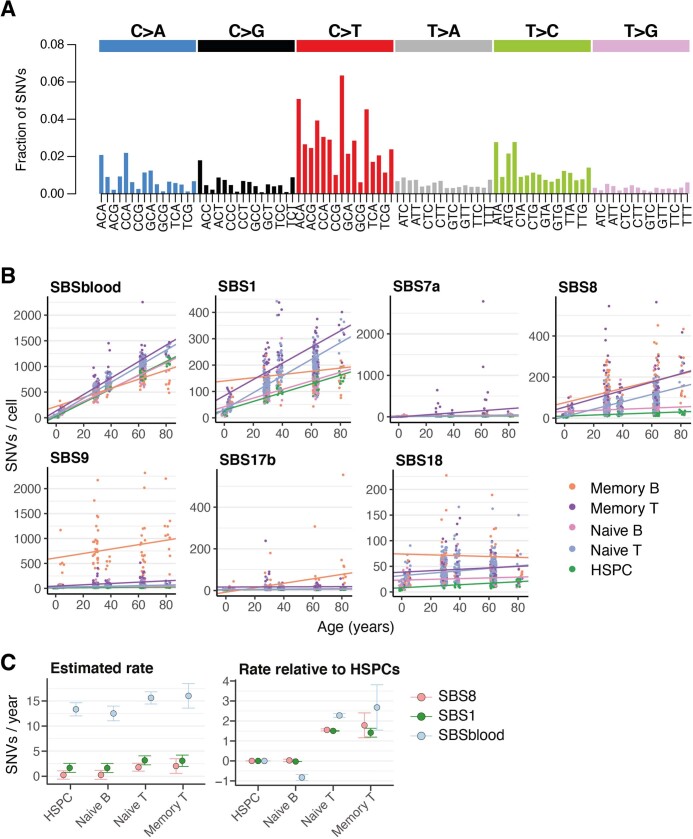
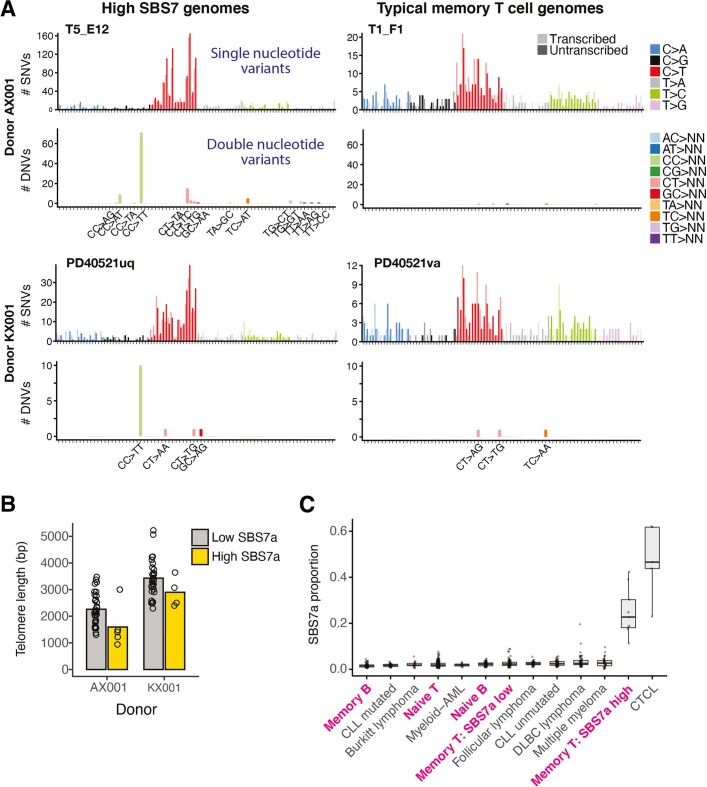
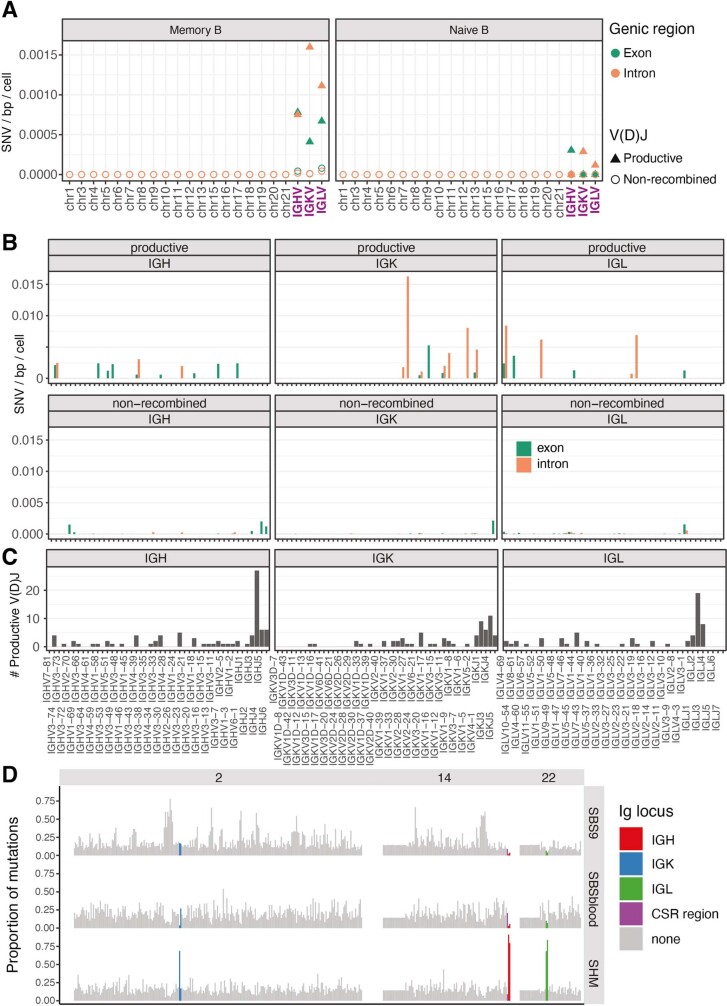
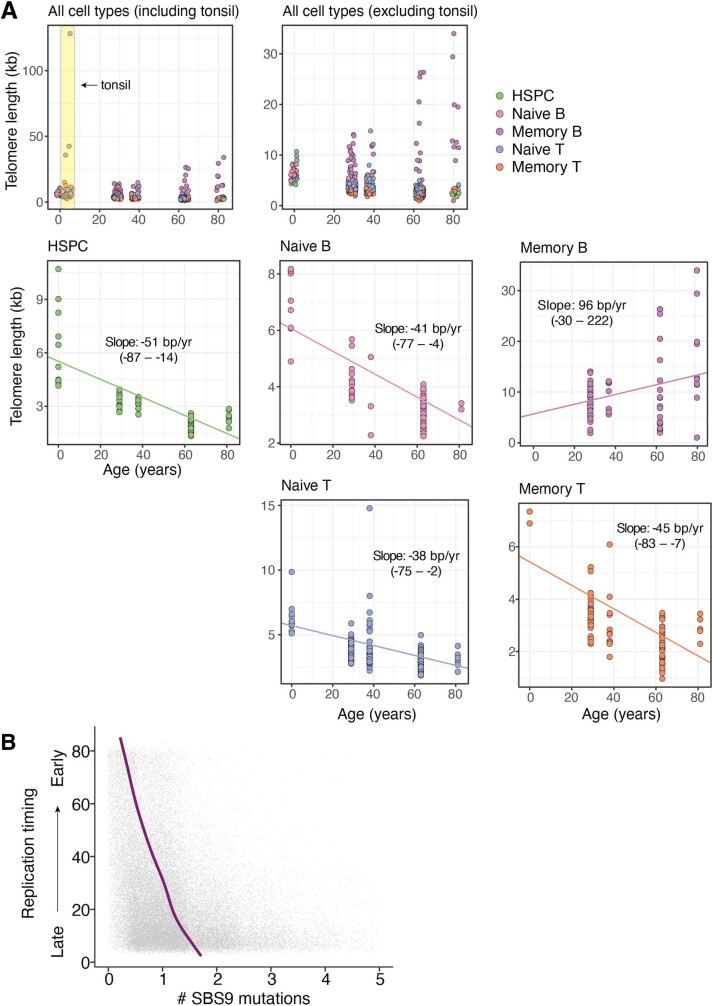
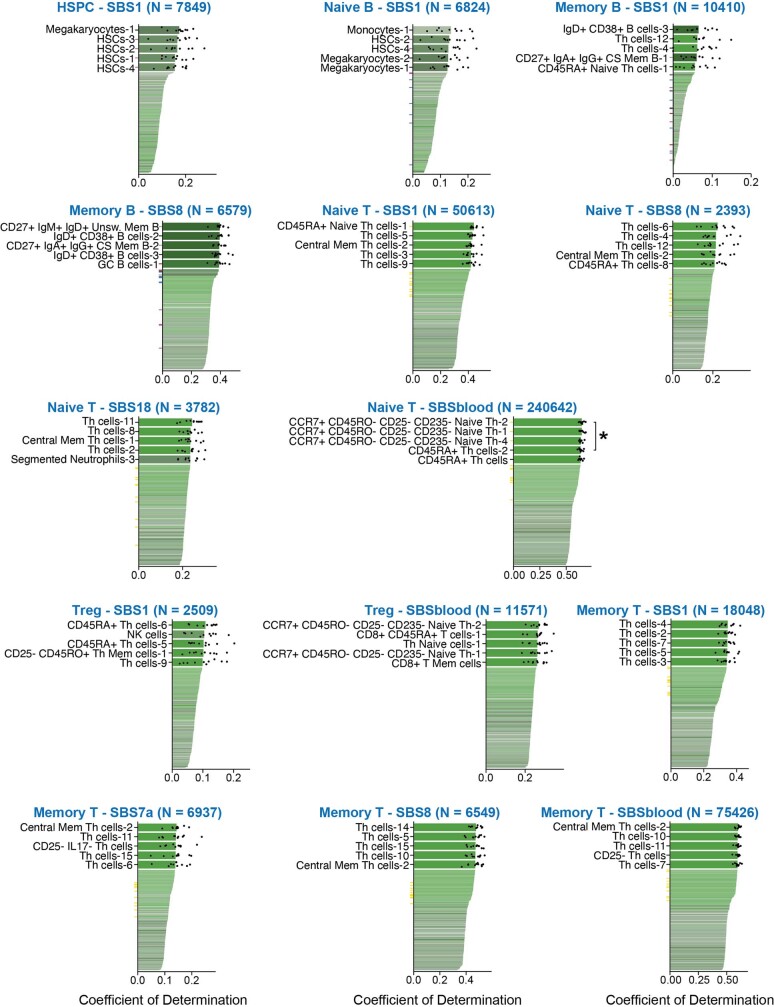
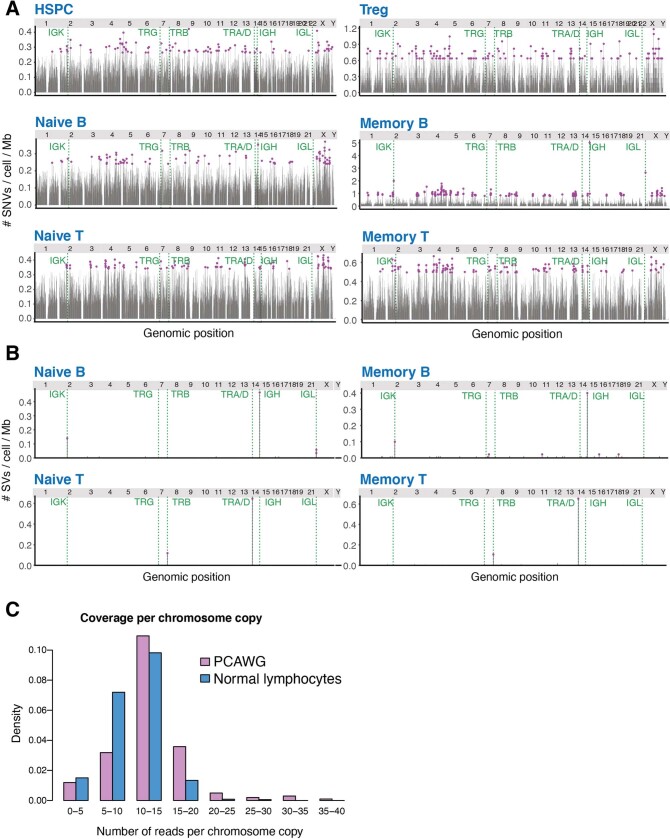
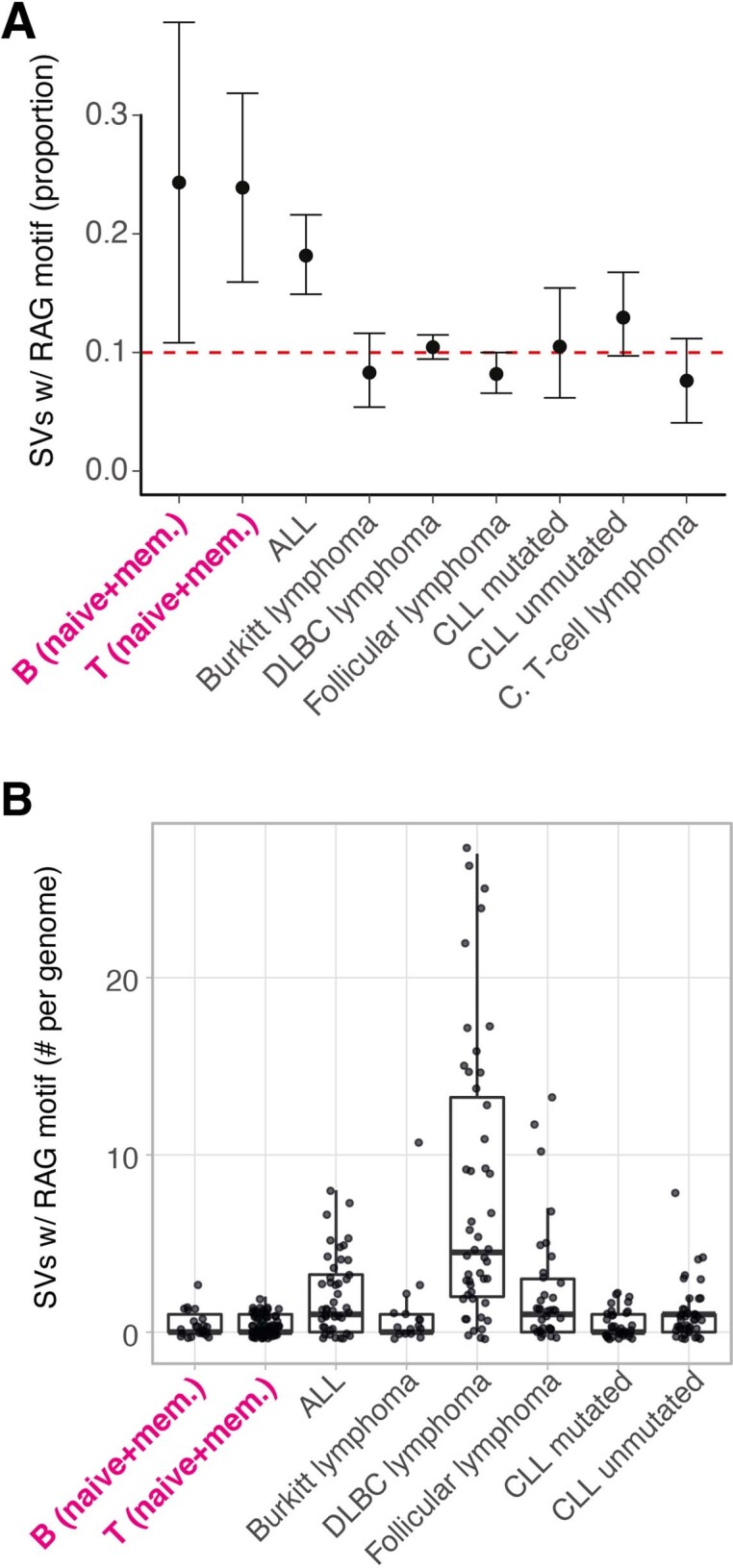
The lymphocyte genome is prone to many threats, including programmed mutation during differentiation1, antigen-driven proliferation and residency in diverse microenvironments. Here, after developing protocols for expansion of single-cell lymphocyte cultures, we sequenced whole genomes from 717 normal naive and memory B and T cells and haematopoietic stem cells. All lymphocyte subsets carried more point mutations and structural variants than haematopoietic stem cells, with higher burdens in memory cells than in naive cells, and with T cells accumulating mutations at a higher rate throughout life. Off-target effects of immunological diversification accounted for approximately half of the additional differentiation-associated mutations in lymphocytes. Memory B cells acquired, on average, 18 off-target mutations genome-wide for every on-target IGHV mutation during the germinal centre reaction. Structural variation was 16-fold higher in lymphocytes than in stem cells, with around 15% of deletions being attributable to off-target recombinase-activating gene activity. DNA damage from ultraviolet light exposure and other sporadic mutational processes generated hundreds to thousands of mutations in some memory cells. The mutation burden and signatures of normal B cells were broadly similar to those seen in many B-cell cancers, suggesting that malignant transformation of lymphocytes arises from the same mutational processes that are active across normal ontogeny. The mutational landscape of normal lymphocytes chronicles the off-target effects of programmed genome engineering during immunological diversification and the consequences of differentiation, proliferation and residency in diverse microenvironments.
© 2022. The Author(s).
Conflict of interest statement
G.G. receives research funds from Pharmacyclics and IBM. G.G. is an inventor on multiple patents related to bioinformatics methods (MuTect, MutSig, ABSOLUTE, MSMutSig, MSMuTect, POLYSOLVER and TensorQTL). G.G. is a founder, consultant and holds privately held equity in Scorpion Therapeutics. D.J.H. receives research funding from AstraZeneca and D.G.K. receives research funding from STRM.bio. All other authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
