

Spatial multi-omic map of human myocardial infarction
- PMID: 35948637
- PMCID: PMC9364862
- DOI: 10.1038/s41586-022-05060-x
Spatial multi-omic map of human myocardial infarction
Abstract
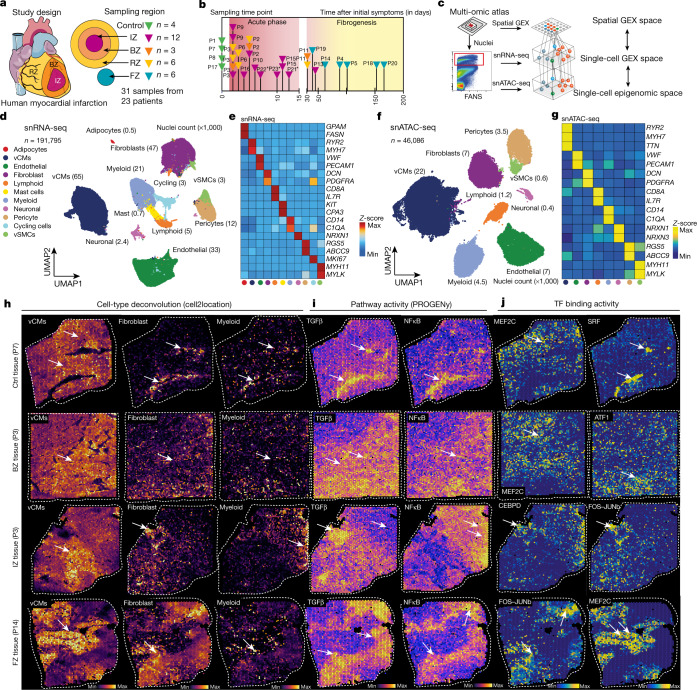
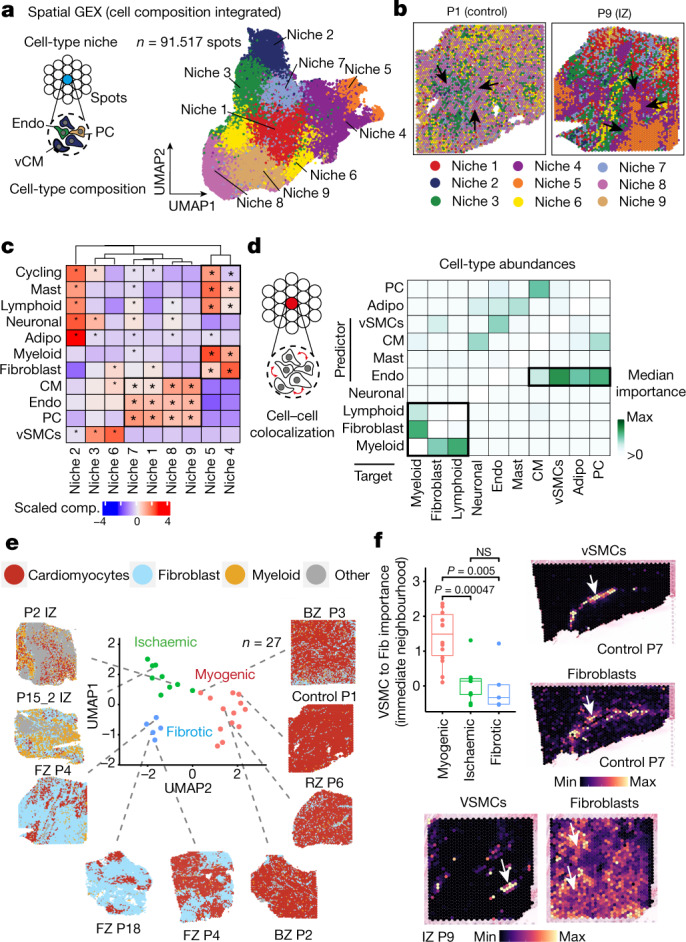
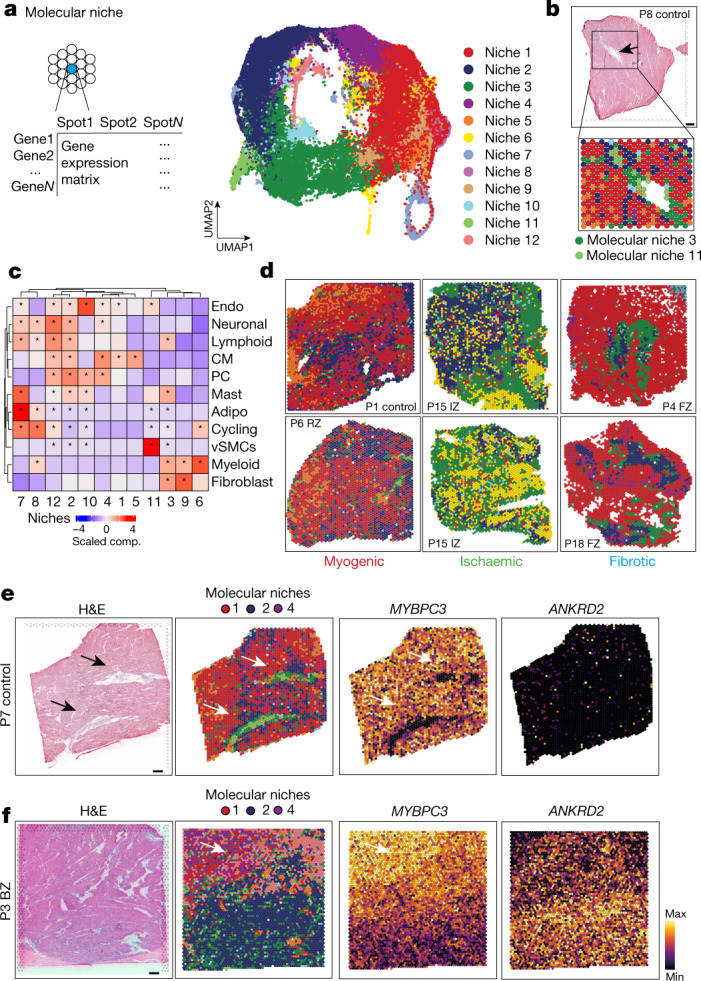
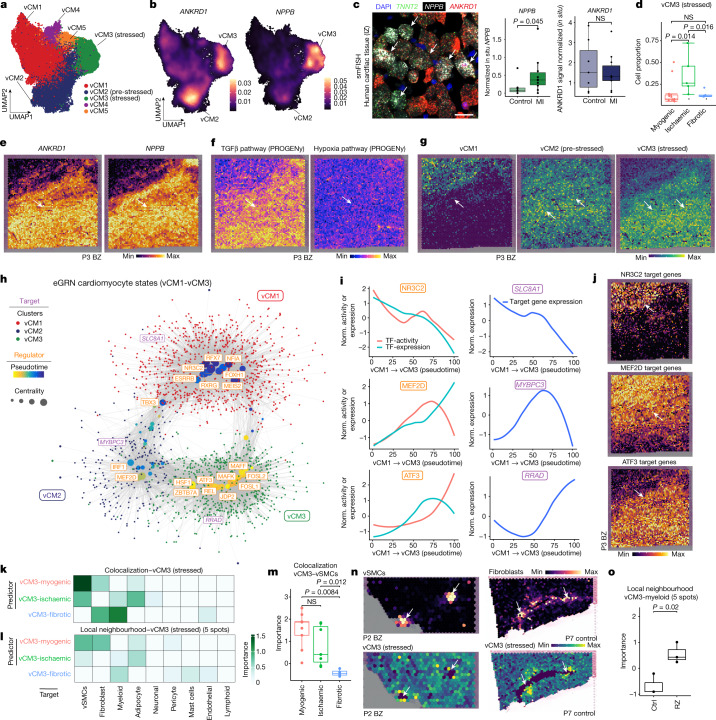
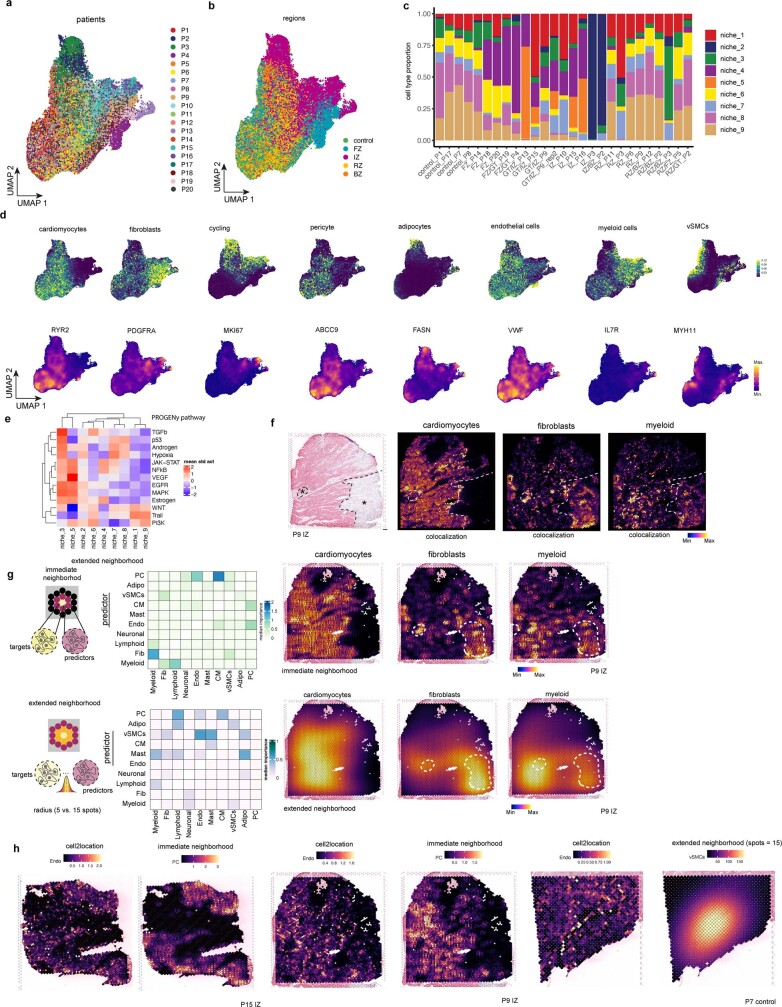
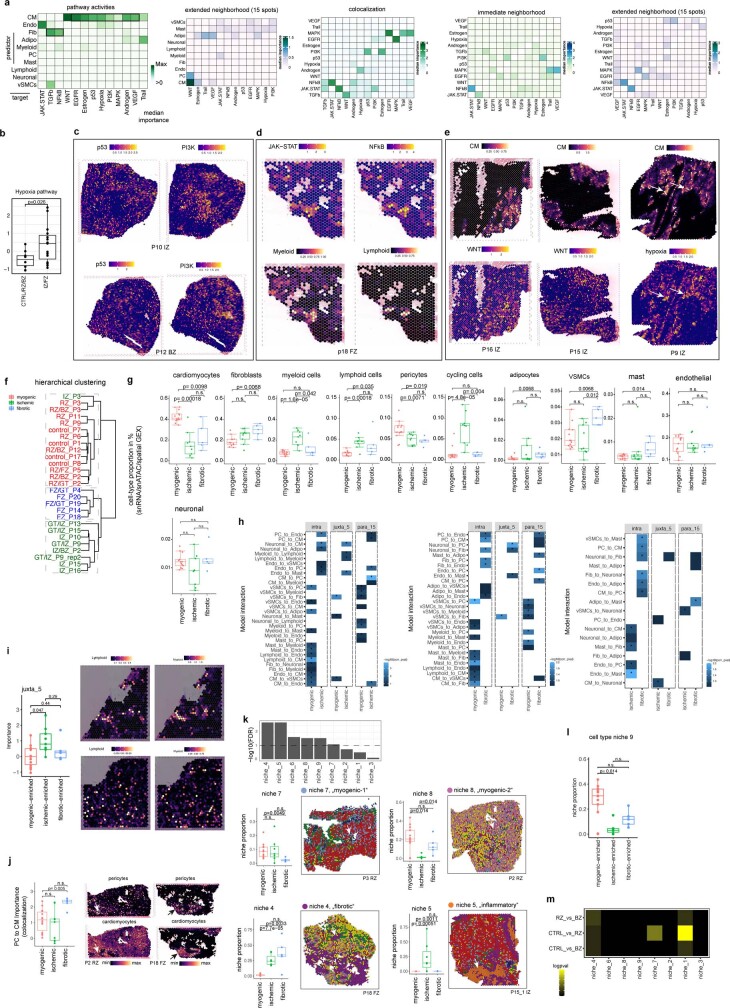
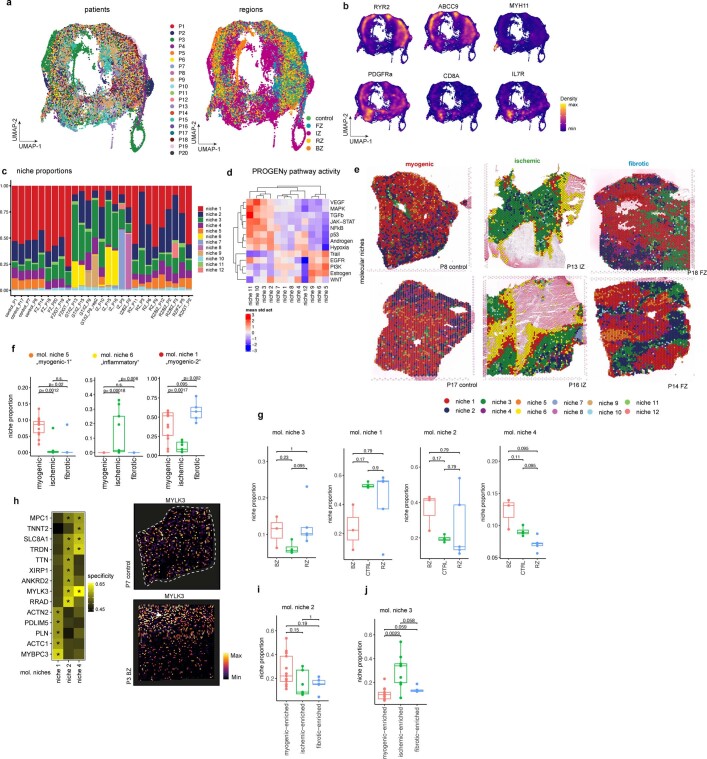
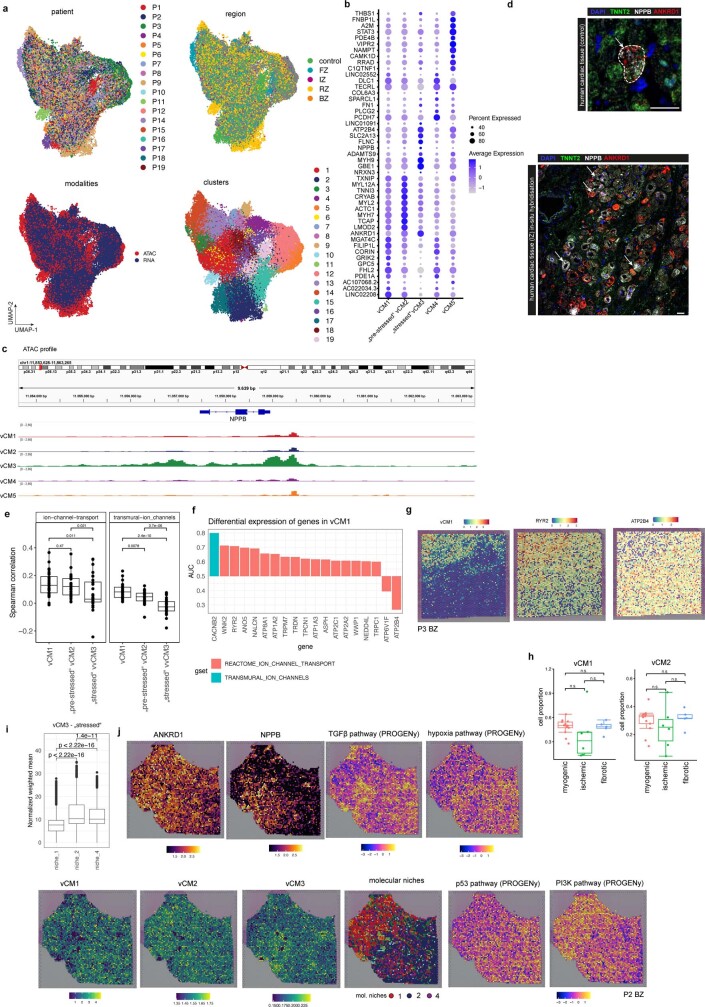
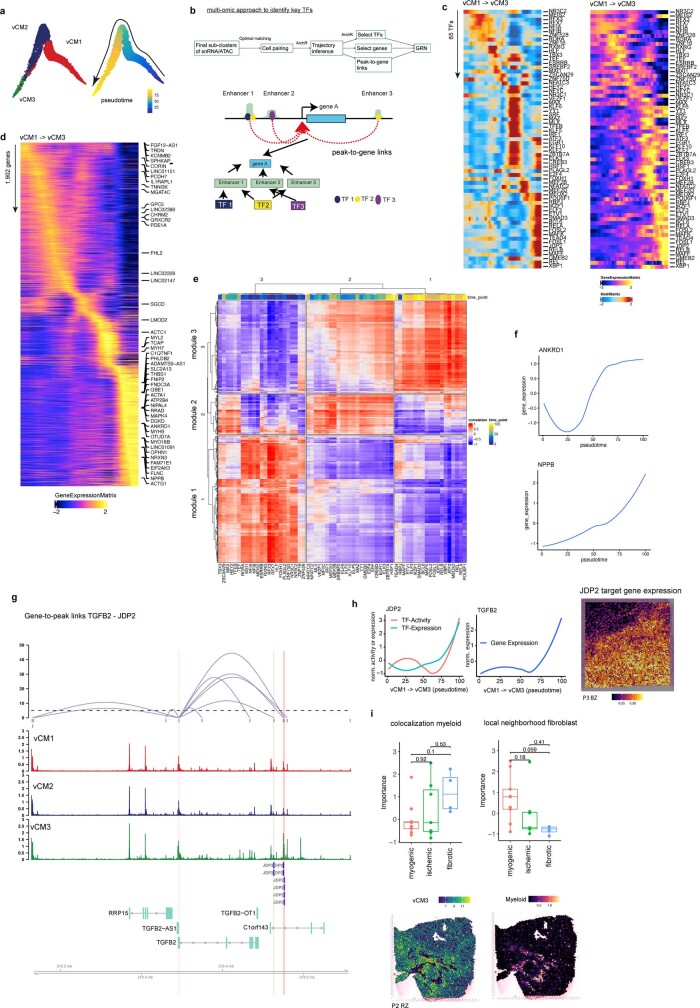
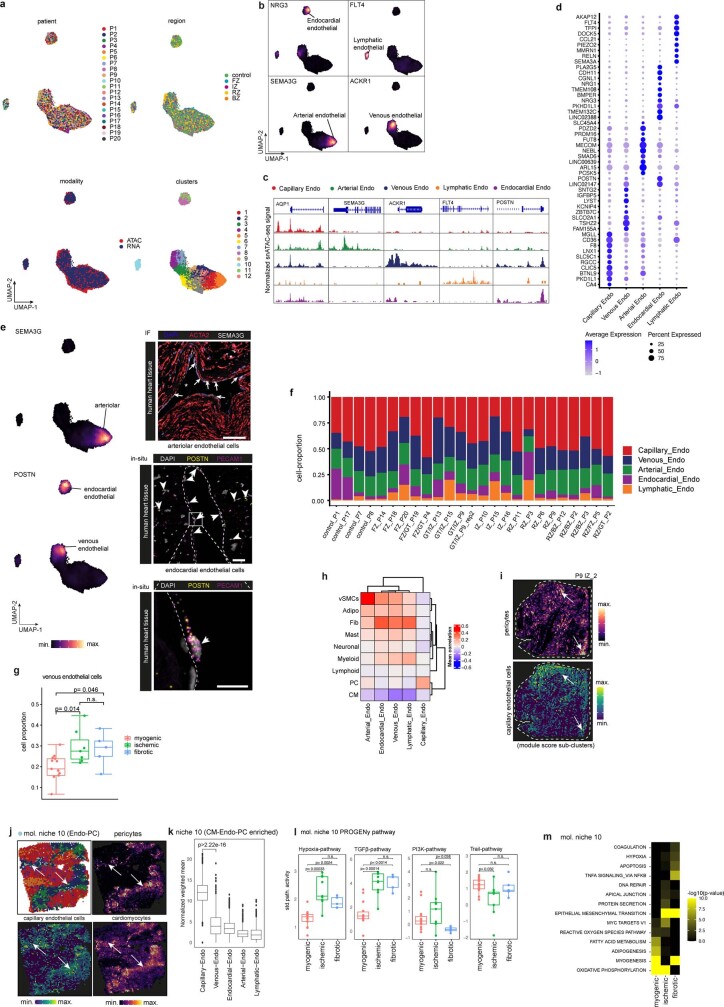
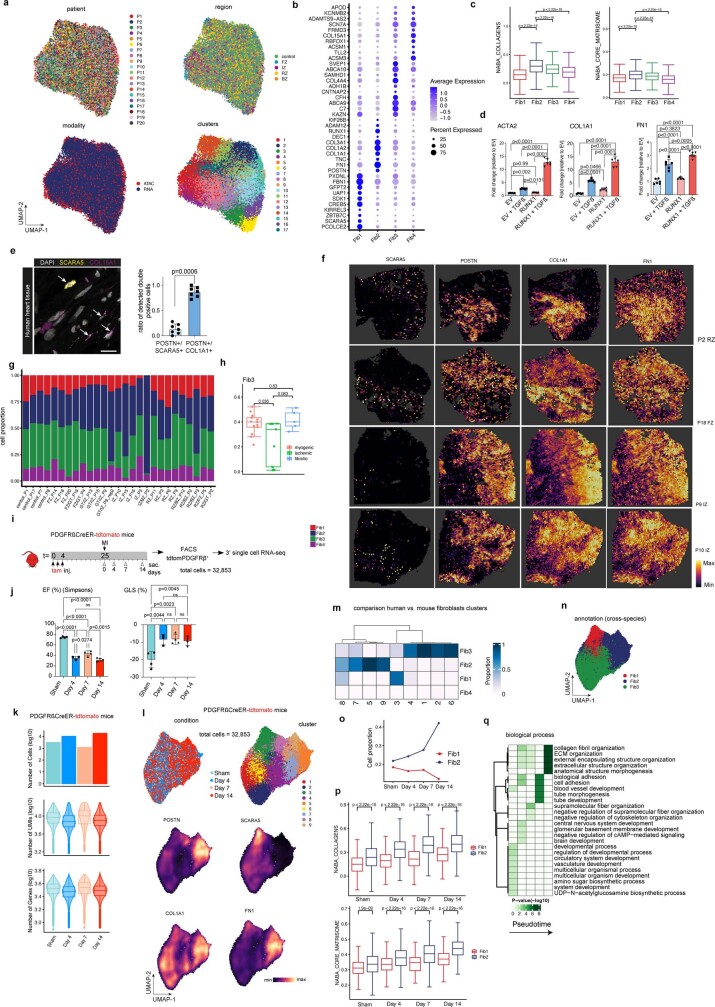
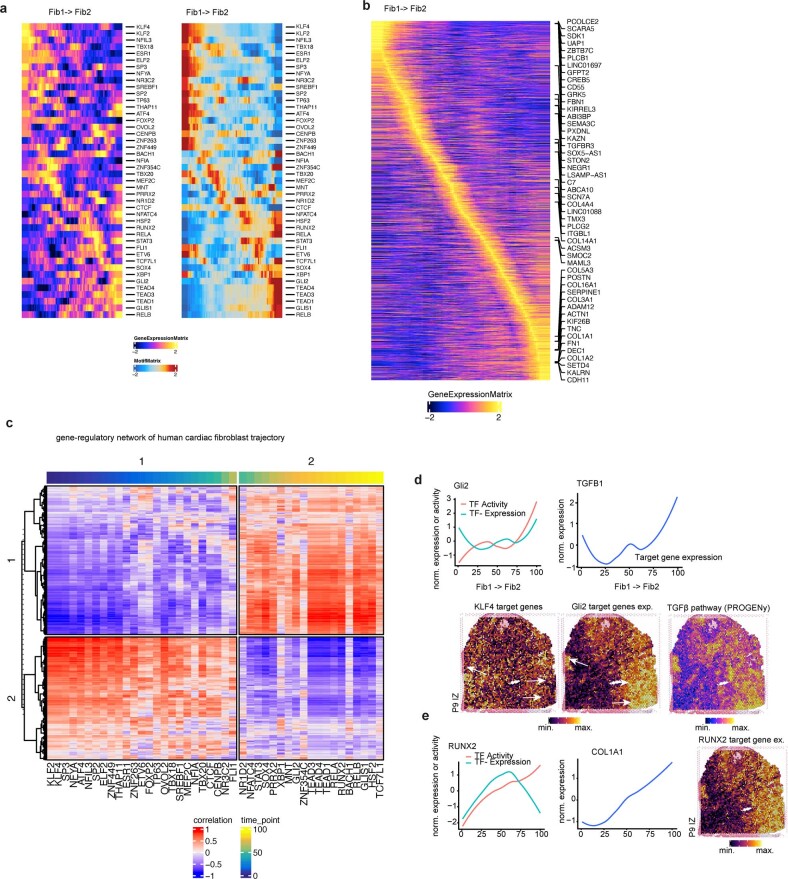
Myocardial infarction is a leading cause of death worldwide1. Although advances have been made in acute treatment, an incomplete understanding of remodelling processes has limited the effectiveness of therapies to reduce late-stage mortality2. Here we generate an integrative high-resolution map of human cardiac remodelling after myocardial infarction using single-cell gene expression, chromatin accessibility and spatial transcriptomic profiling of multiple physiological zones at distinct time points in myocardium from patients with myocardial infarction and controls. Multi-modal data integration enabled us to evaluate cardiac cell-type compositions at increased resolution, yielding insights into changes of the cardiac transcriptome and epigenome through the identification of distinct tissue structures of injury, repair and remodelling. We identified and validated disease-specific cardiac cell states of major cell types and analysed them in their spatial context, evaluating their dependency on other cell types. Our data elucidate the molecular principles of human myocardial tissue organization, recapitulating a gradual cardiomyocyte and myeloid continuum following ischaemic injury. In sum, our study provides an integrative molecular map of human myocardial infarction, represents an essential reference for the field and paves the way for advanced mechanistic and therapeutic studies of cardiac disease.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests directly related to this work. The authors however disclose unrelated funding, honorariums and ownership as follows: R.K. has grants from Travere Therapeutics, Galapagos, Chugai and Novo Nordisk and is a consultant for Bayer, Pfizer, Novo Nordisk and Gruenenthal. J.S.-R. reports funding from GSK and Sanofi and fees from Travere Therapeutics and Astex Therapeutics. K.L. has grants from Novartis and Amgen and receives consulting fees from Medtronic and Implicit Biosciences. I.G.C. has a grant from Illumina. L.S. has received grants from Bayer, Boehringer Ingelheim and Nattopharma, is consultant for ImmunoDiagnostics Systems (IDS) and is a shareholder in Coagulation Profile. L.W.V.L. reports consultancy fees to UMCU from Abbott, Medtronic, Vifor and Novartis.
Figures
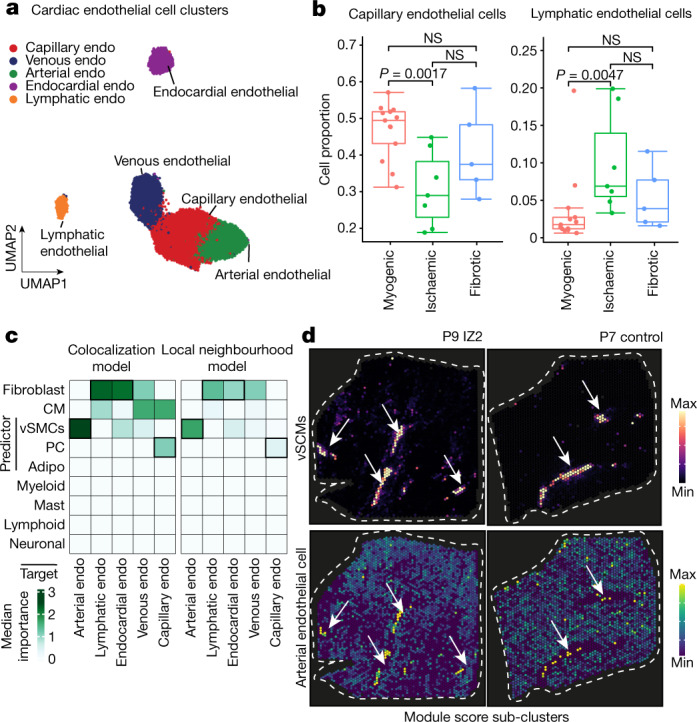
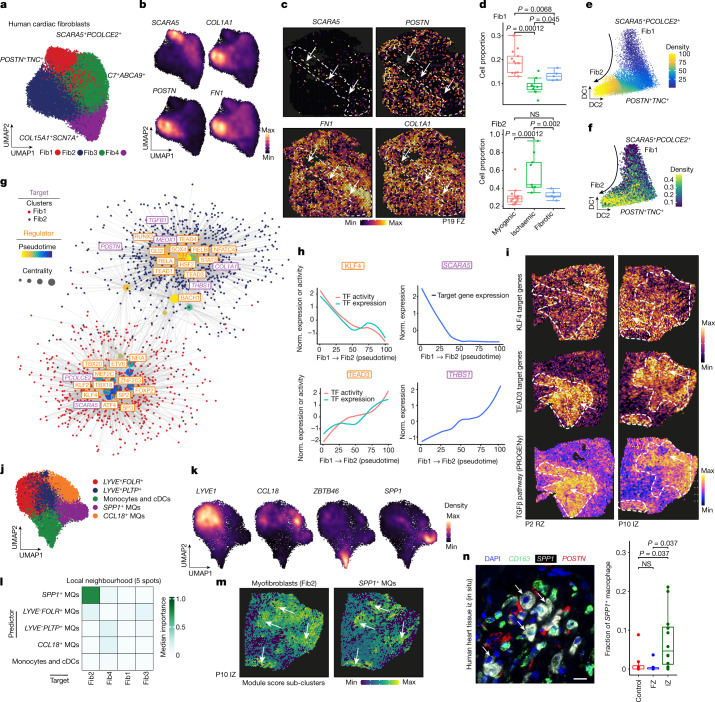
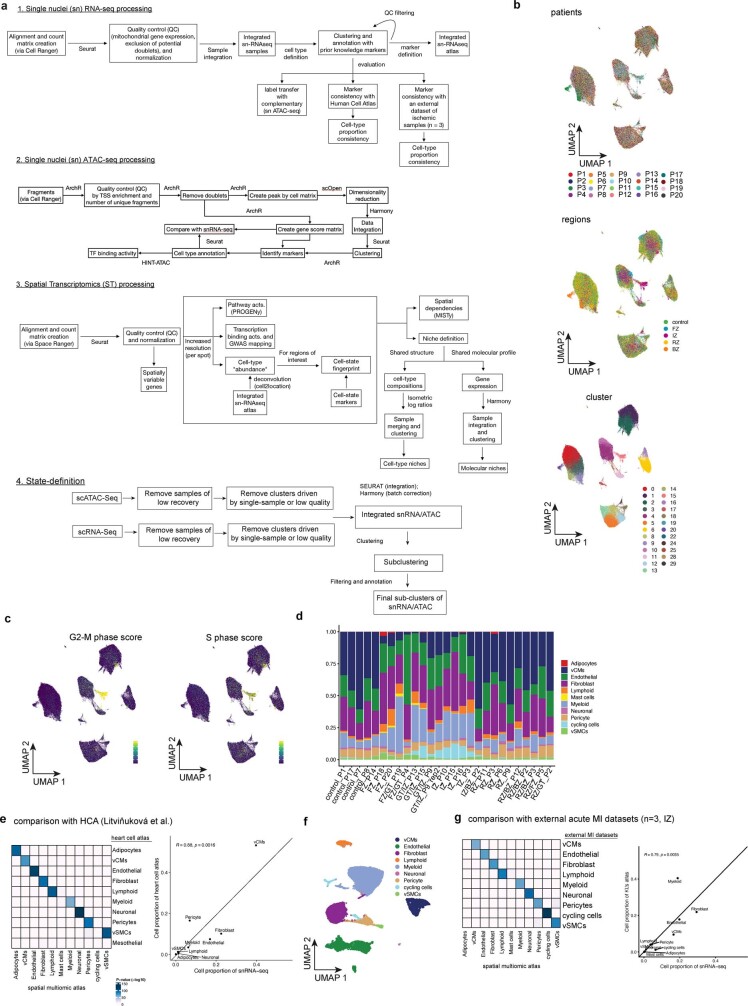
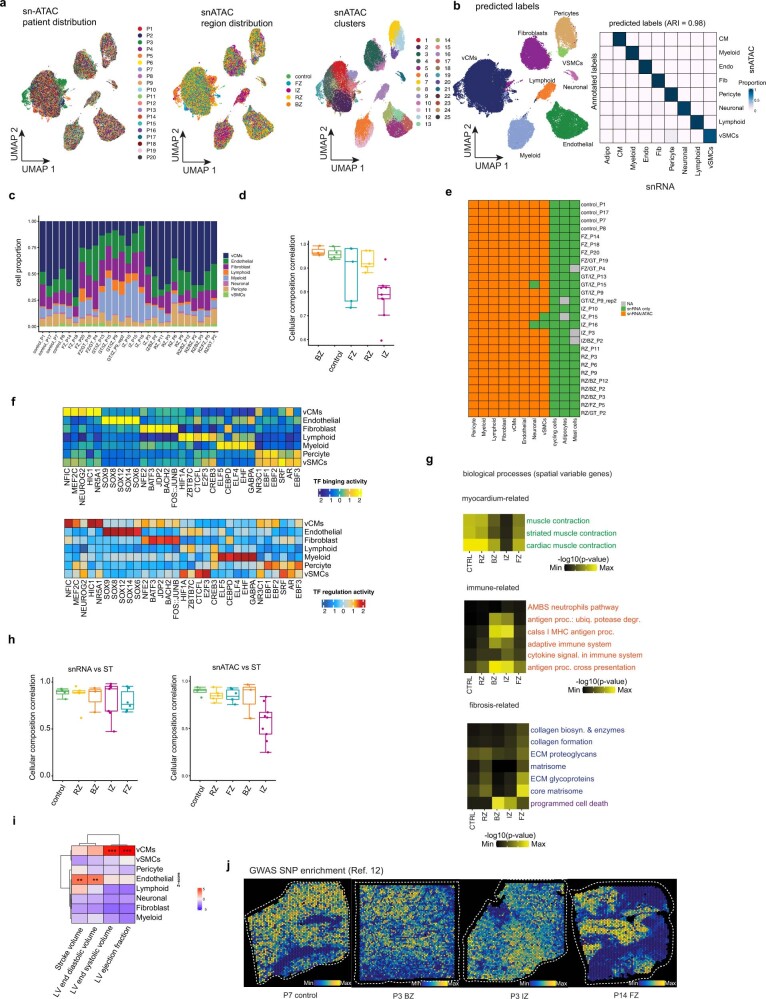
Comment in
-
Mind the map: a multi-omic approach to post-ischaemic cardiac remodelling paves the way for future cardiovascular research.Eur Heart J. 2022 Dec 1;43(45):4677-4678. doi: 10.1093/eurheartj/ehac525. Eur Heart J. 2022. PMID: 36214046 No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
