

Spatially resolved clonal copy number alterations in benign and malignant tissue
- PMID: 35948708
- PMCID: PMC9365699
- DOI: 10.1038/s41586-022-05023-2
Spatially resolved clonal copy number alterations in benign and malignant tissue
Abstract
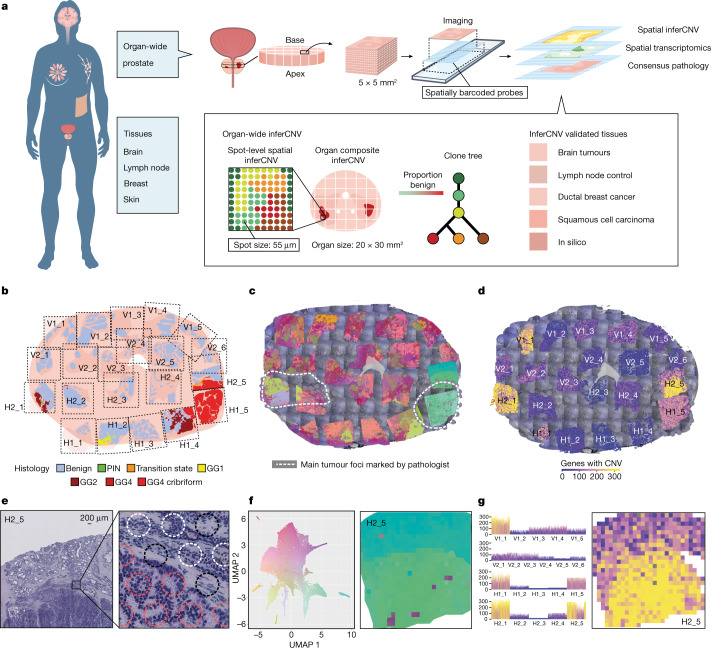
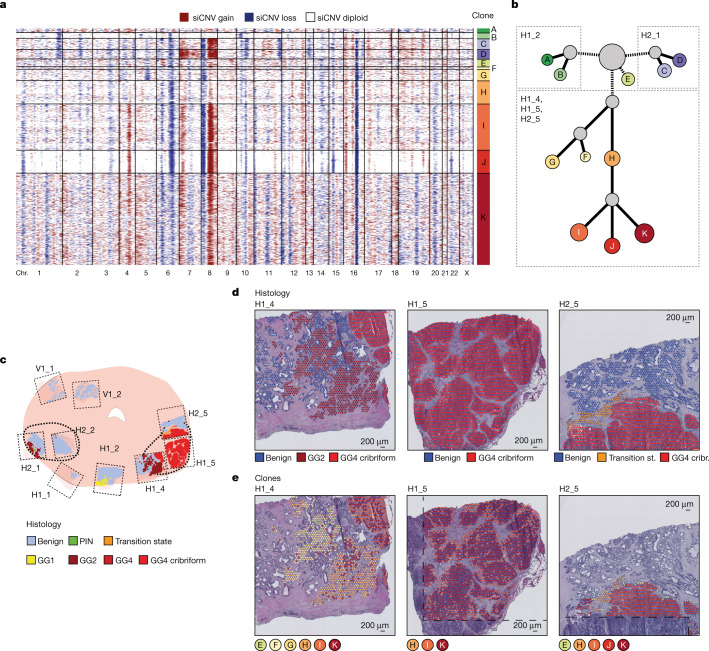
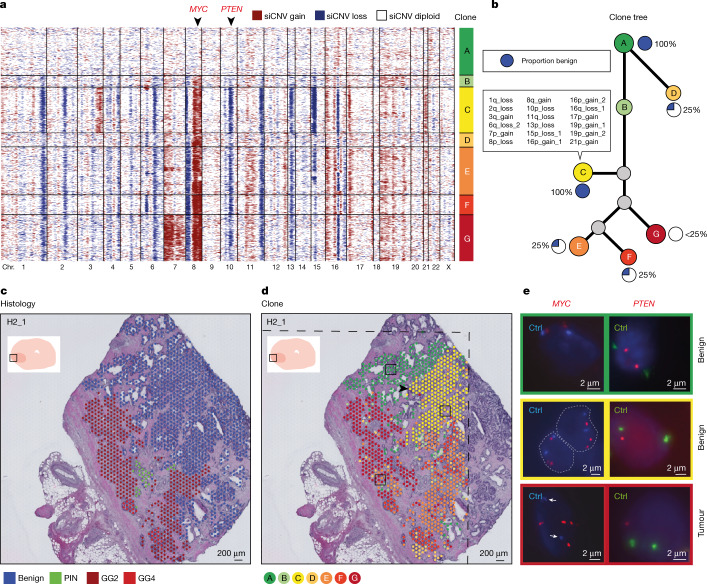
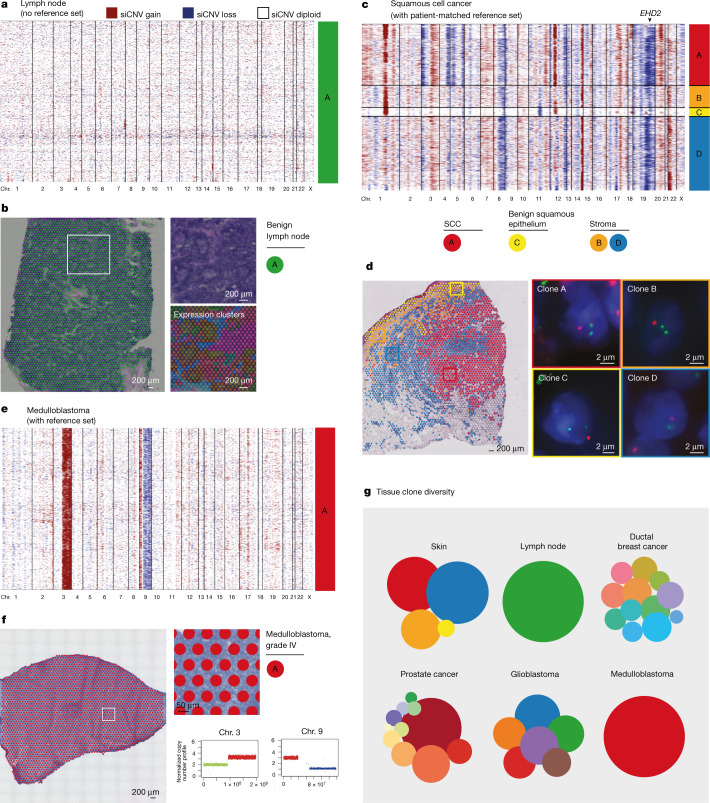
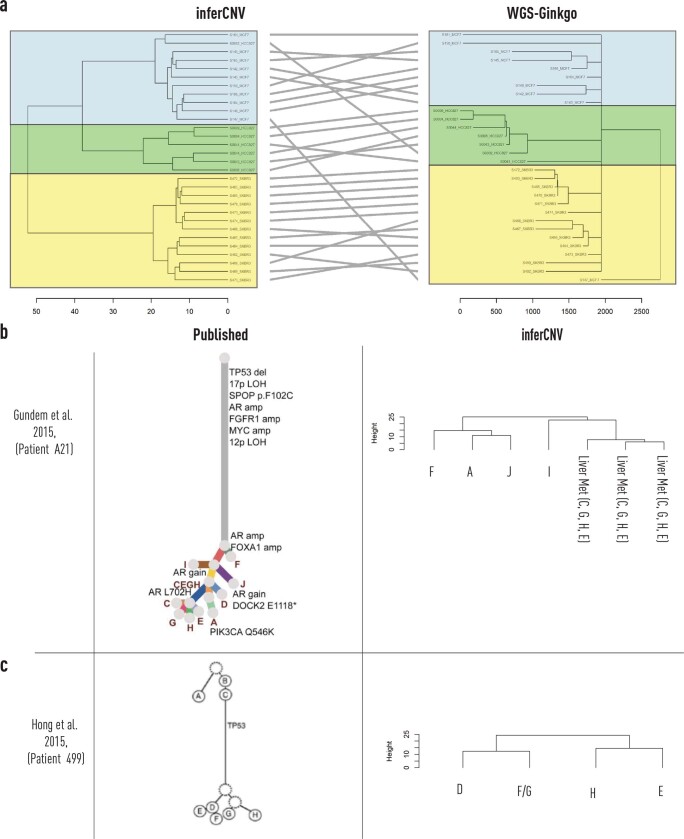
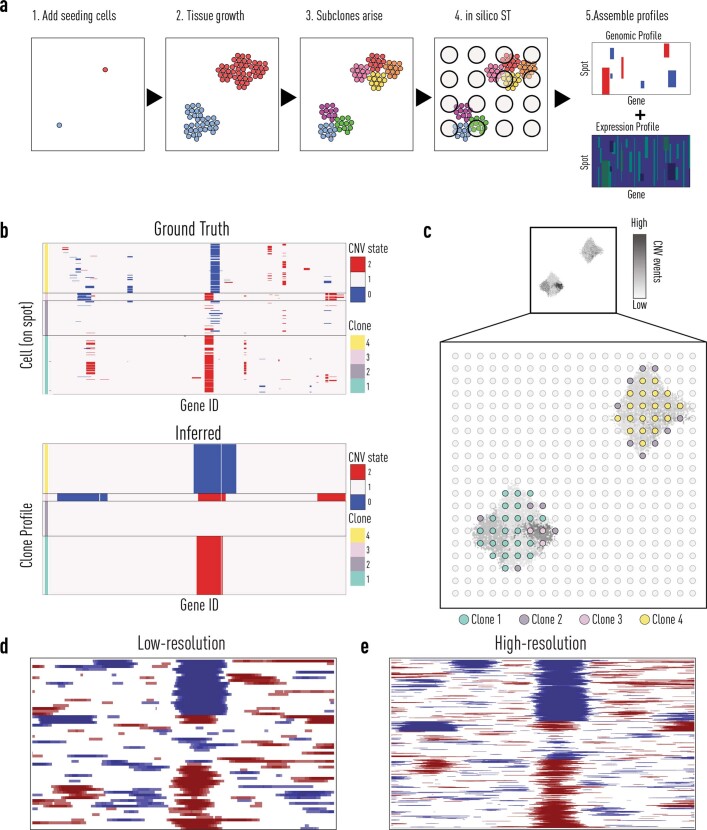
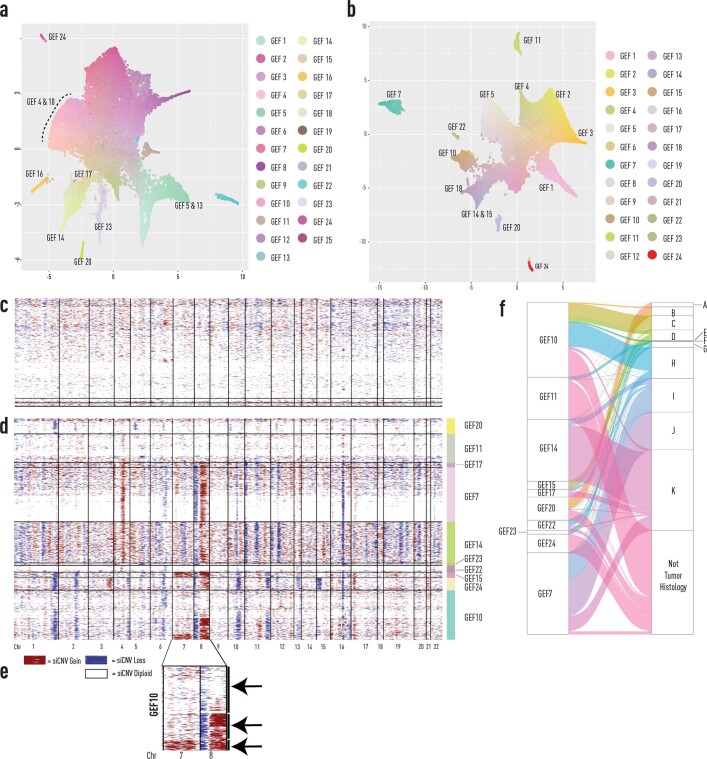
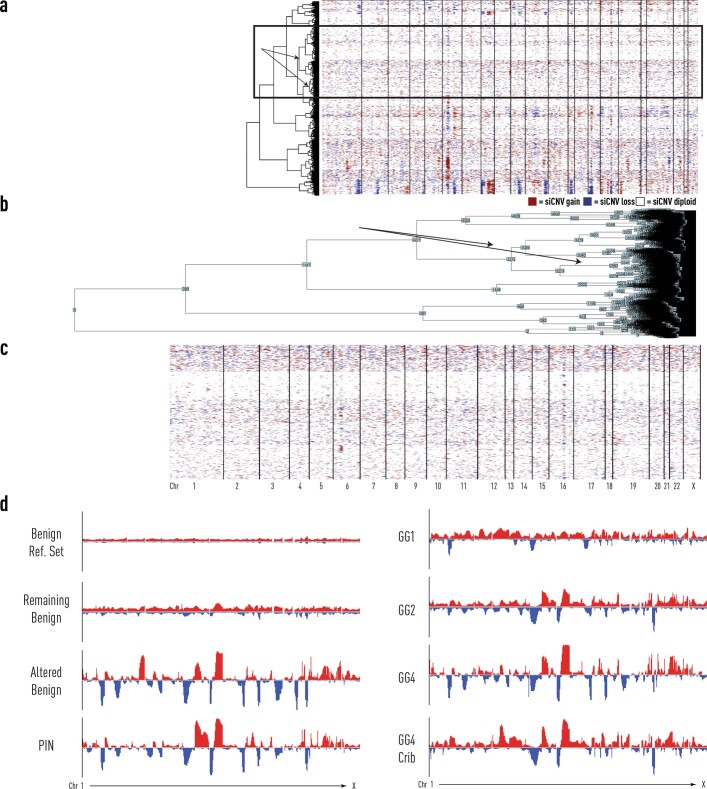
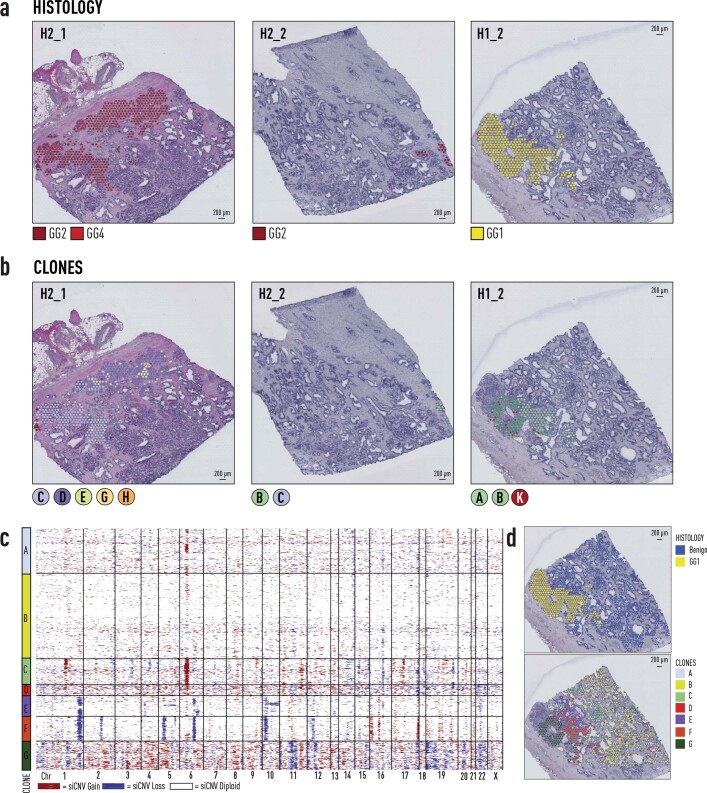
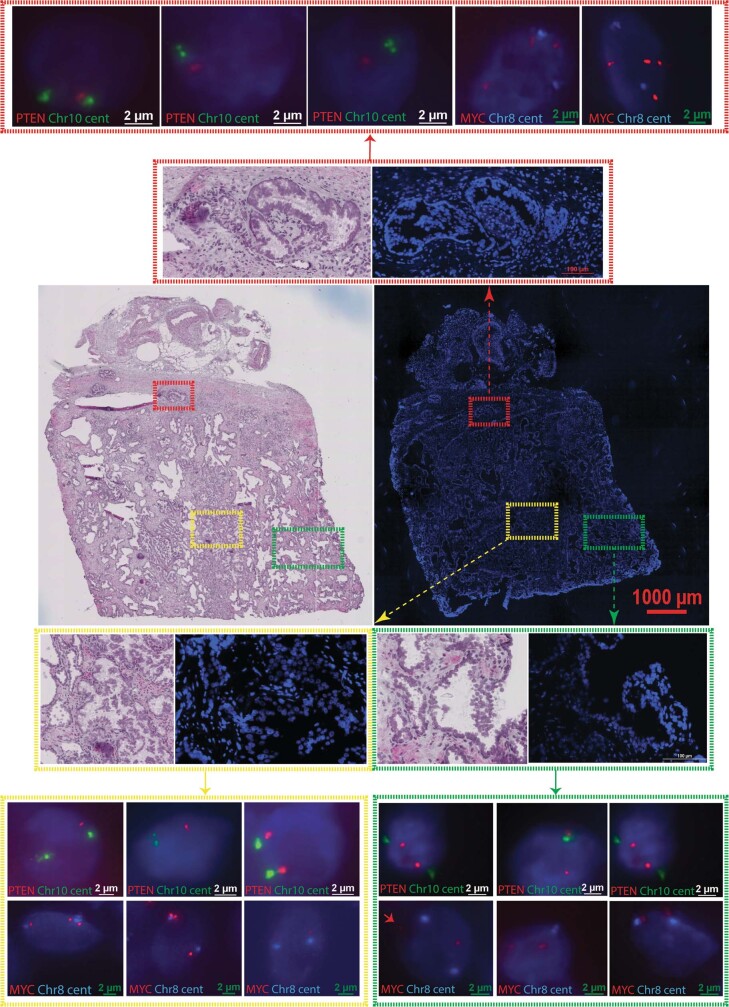
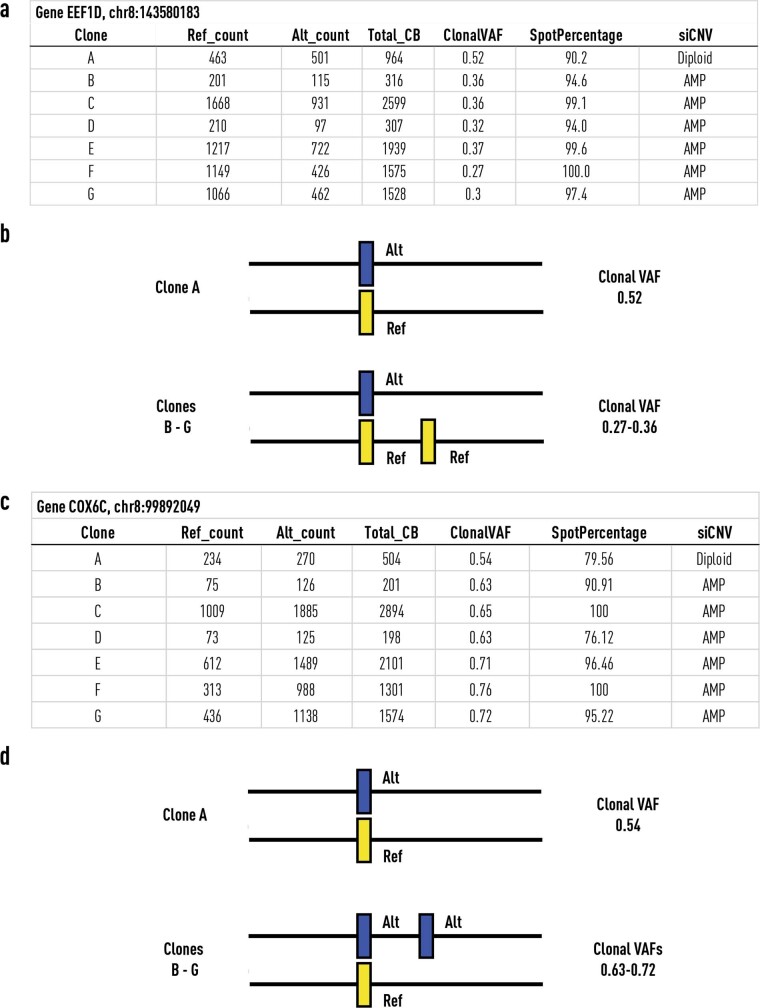
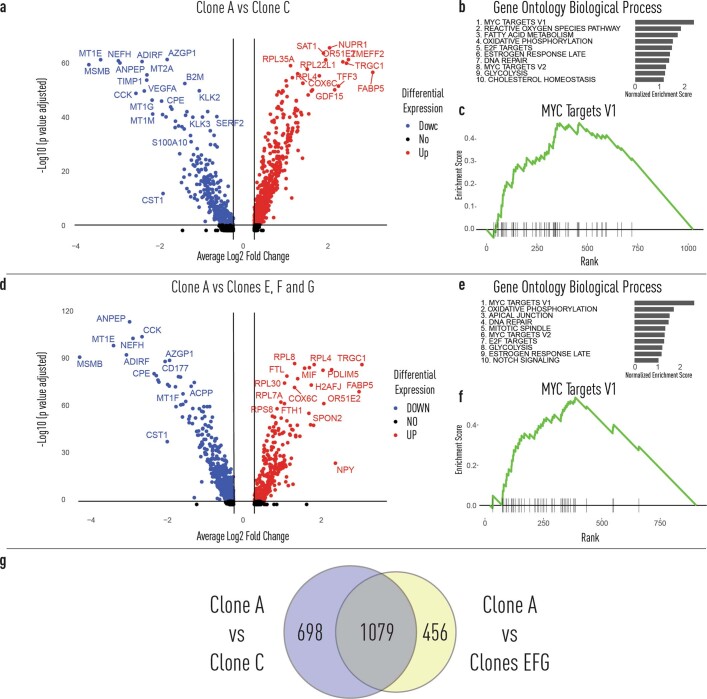
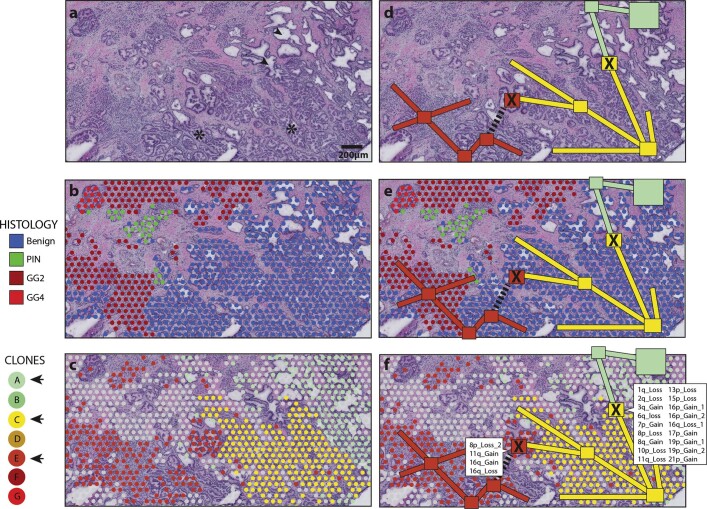
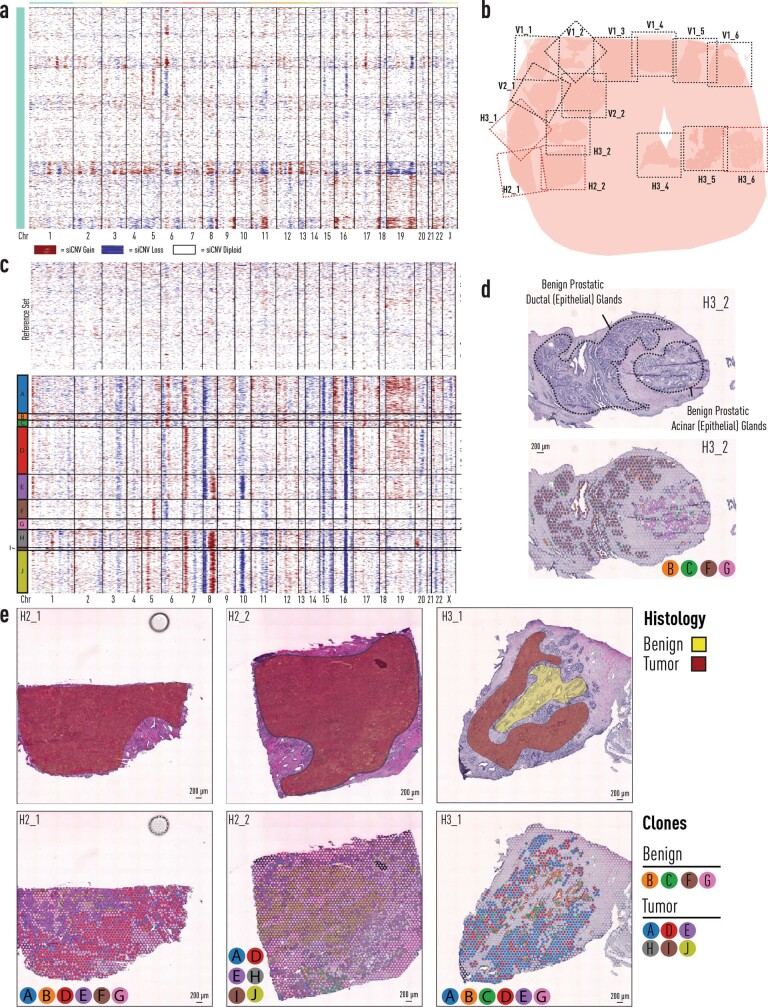
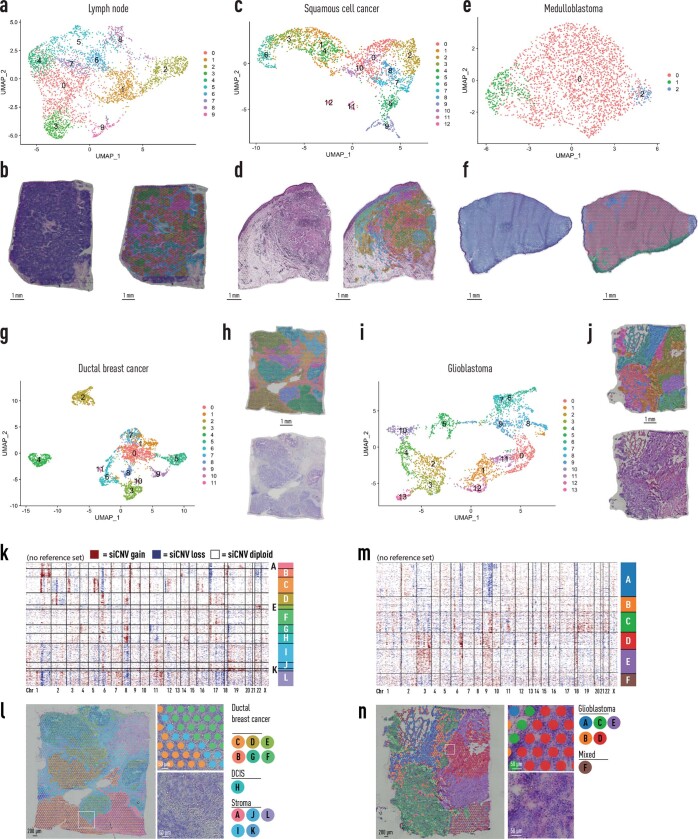
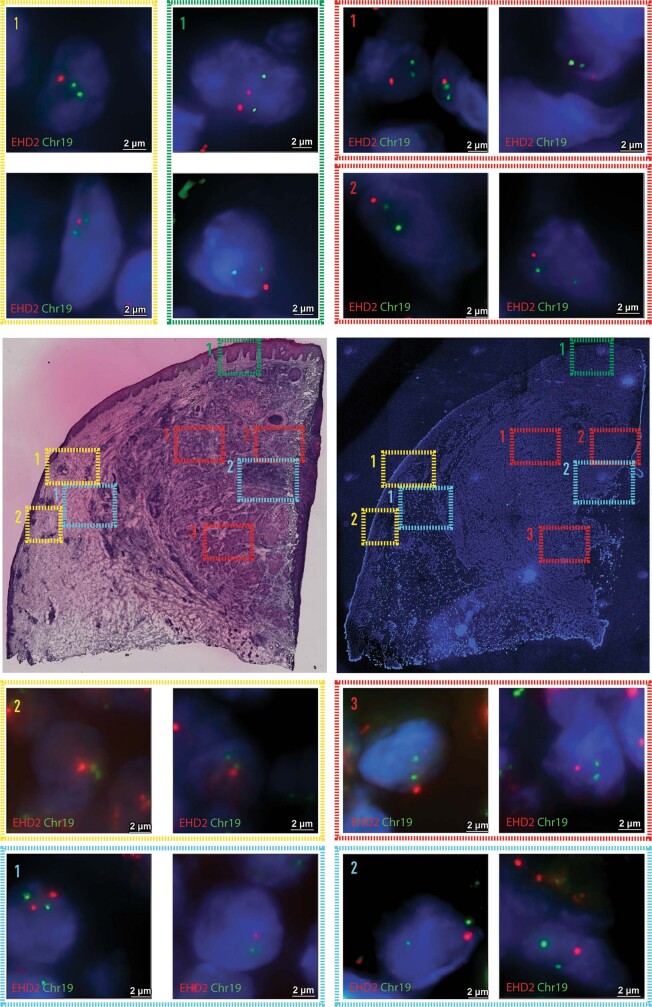
Defining the transition from benign to malignant tissue is fundamental to improving early diagnosis of cancer1. Here we use a systematic approach to study spatial genome integrity in situ and describe previously unidentified clonal relationships. We used spatially resolved transcriptomics2 to infer spatial copy number variations in >120,000 regions across multiple organs, in benign and malignant tissues. We demonstrate that genome-wide copy number variation reveals distinct clonal patterns within tumours and in nearby benign tissue using an organ-wide approach focused on the prostate. Our results suggest a model for how genomic instability arises in histologically benign tissue that may represent early events in cancer evolution. We highlight the power of capturing the molecular and spatial continuums in a tissue context and challenge the rationale for treatment paradigms, including focal therapy.
© 2022. The Author(s).
Conflict of interest statement
M.H., M.M., R.M., L.K., A.A., L.L., L.A.G., K.T. and J.L. are scientific consultants to 10x Genomics, Inc.
Figures

Comment in
-
Re: Spatially Resolved Clonal Copy Number Alterations in Benign and Malignant Tissue.Eur Urol. 2023 Feb;83(2):183. doi: 10.1016/j.eururo.2022.11.013. Epub 2022 Dec 1. Eur Urol. 2023. PMID: 36464529 No abstract available.
References
-
- Chen JZ, Gokden N, Greene GF, Mukunyadzi P, Kadlubar FF. Extensive somatic mitochondrial mutations in primary prostate cancer using laser capture microdissection. Cancer Res. 2002;62:6470–6474. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
