

Liver transplantation for late-onset ornithine transcarbamylase deficiency: A case report
- PMID: 35949846
- PMCID: PMC9254178
- DOI: 10.12998/wjcc.v10.i18.6156
Liver transplantation for late-onset ornithine transcarbamylase deficiency: A case report
Abstract
Background: Ornithine transcarbamylase deficiency (OTCD) is an X-linked inherited disorder and characterized by marked elevation of blood ammonia. The goal of treatment is to minimize the neurological damage caused by hyperammonemia. OTCD can be cured by liver transplantation (LT). Post-transplant patients can discontinue anti- hyperammonemia agents and consume a regular diet without the risk of developing hyperammonemia. The neurological damage caused by hyperammonemia is almost irreversible.
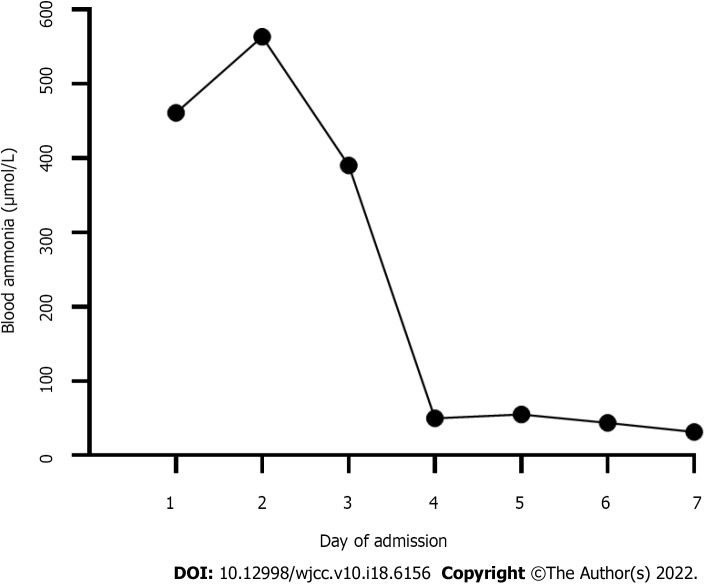
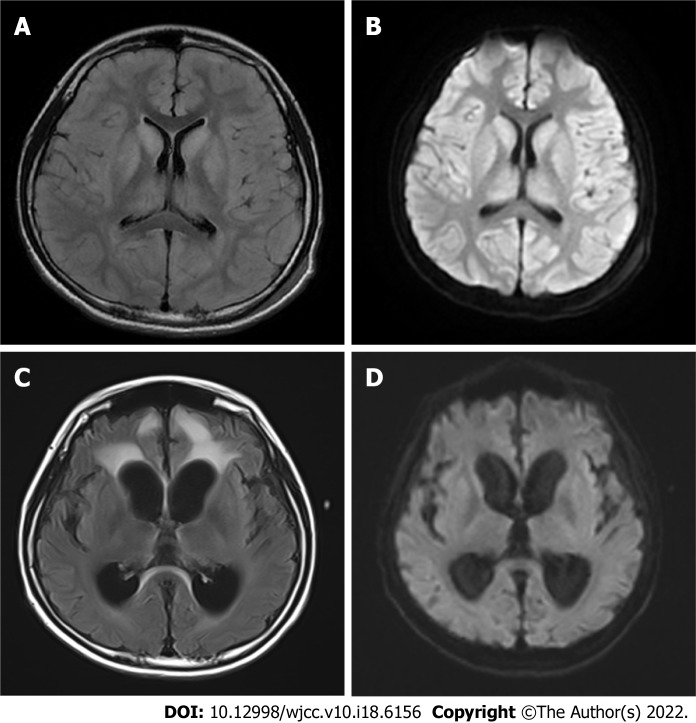
Case summary: An 11.7-year-old boy presented with headache, vomiting, and altered consciousness. The patient was diagnosed with late-onset OTCD. After nitrogen scavenging treatment and a protein-free diet, ammonia levels were reduced to normal on the third day of admission. Nevertheless, the patient remained in a moderate coma. After discussion, LT was performed. Following LT, the patient's blood ammonia and biochemical indicators stabilized in the normal range, he regained consciousness, and his nervous system function significantly recovered. Two months after LT, blood amino acids and urine organic acids were normal, and brain magnetic resonance imaging showed a decrease in subcortical lesions.
Conclusion: LT can significantly improve partial neurological impairment caused by late-onset OTCD hyperammonemic encephalopathy, and LT can be actively considered when early drug therapy is ineffective.
Keywords: Case report; Hyperammonemic encephalopathy; Liver transplantation; Ornithine transcarbamylase deficiency; Urea cycle disorder.
©The Author(s) 2022. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Liver transplantation in rare late-onset ornithine transcarbamylase deficiency with central nervous system injury: A case report and review of the literature.Brain Behav. 2022 Oct;12(10):e2765. doi: 10.1002/brb3.2765. Epub 2022 Sep 20. Brain Behav. 2022. PMID: 36128655 Free PMC article. Review.
-
Late-Onset Ornithine Transcarbamylase Deficiency Complicated with Extremely High Serum Ammonia Level: Prompt Induction of Hemodialysis as the Key to Successful Treatment.Am J Case Rep. 2022 Nov 15;23:e937658. doi: 10.12659/AJCR.937658. Am J Case Rep. 2022. PMID: 36377209 Free PMC article.
-
Liver transplantation in ornithine transcarbamylase deficiency: A retrospective multicentre cohort study.Mol Genet Metab Rep. 2023 Nov 5;37:101020. doi: 10.1016/j.ymgmr.2023.101020. eCollection 2023 Dec. Mol Genet Metab Rep. 2023. PMID: 38053940 Free PMC article.
-
Complete recovery from acute encephalopathy of late-onset ornithine transcarbamylase deficiency in a 3-year-old boy.J Inherit Metab Dis. 2007 Nov;30(6):981. doi: 10.1007/s10545-007-0692-x. Epub 2007 Oct 5. J Inherit Metab Dis. 2007. PMID: 17922216
-
[Consensus on diagnosis and treatment of ornithine trans-carbamylase deficiency].Zhejiang Da Xue Xue Bao Yi Xue Ban. 2020 Oct 25;49(5):539-547. doi: 10.3785/j.issn.1008-9292.2020.04.11. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2020. PMID: 33210478 Free PMC article. Review. Chinese.
Cited by
-
In-depth analysis of OTC A208T case induced by OTC gene mutation and research on the prediction and simulation of the impact on protein function.Front Pediatr. 2024 Sep 12;12:1450859. doi: 10.3389/fped.2024.1450859. eCollection 2024. Front Pediatr. 2024. PMID: 39328593 Free PMC article.
References
-
- Wilcken B. Problems in the management of urea cycle disorders. Mol Genet Metab. 2004;81 Suppl 1:S86–S91. - PubMed
-
- Dionisi-Vici C, Rizzo C, Burlina AB, Caruso U, Sabetta G, Uziel G, Abeni D. Inborn errors of metabolism in the Italian pediatric population: a national retrospective survey. J Pediatr. 2002;140:321–327. - PubMed
-
- Kido J, Nakamura K, Mitsubuchi H, Ohura T, Takayanagi M, Matsuo M, Yoshino M, Shigematsu Y, Yorifuji T, Kasahara M, Horikawa R, Endo F. Long-term outcome and intervention of urea cycle disorders in Japan. J Inherit Metab Dis. 2012;35:777–785. - PubMed
-
- Häberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Lindner M, Mandel H, Martinelli D, Pintos-Morell G, Santer R, Skouma A, Servais A, Tal G, Rubio V, Huemer M, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J Inherit Metab Dis. 2019;42:1192–1230. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
