

The complexity landscape of viral genomes
- PMID: 35950839
- PMCID: PMC9366995
- DOI: 10.1093/gigascience/giac079
The complexity landscape of viral genomes
Abstract
Background: Viruses are among the shortest yet highly abundant species that harbor minimal instructions to infect cells, adapt, multiply, and exist. However, with the current substantial availability of viral genome sequences, the scientific repertory lacks a complexity landscape that automatically enlights viral genomes' organization, relation, and fundamental characteristics.
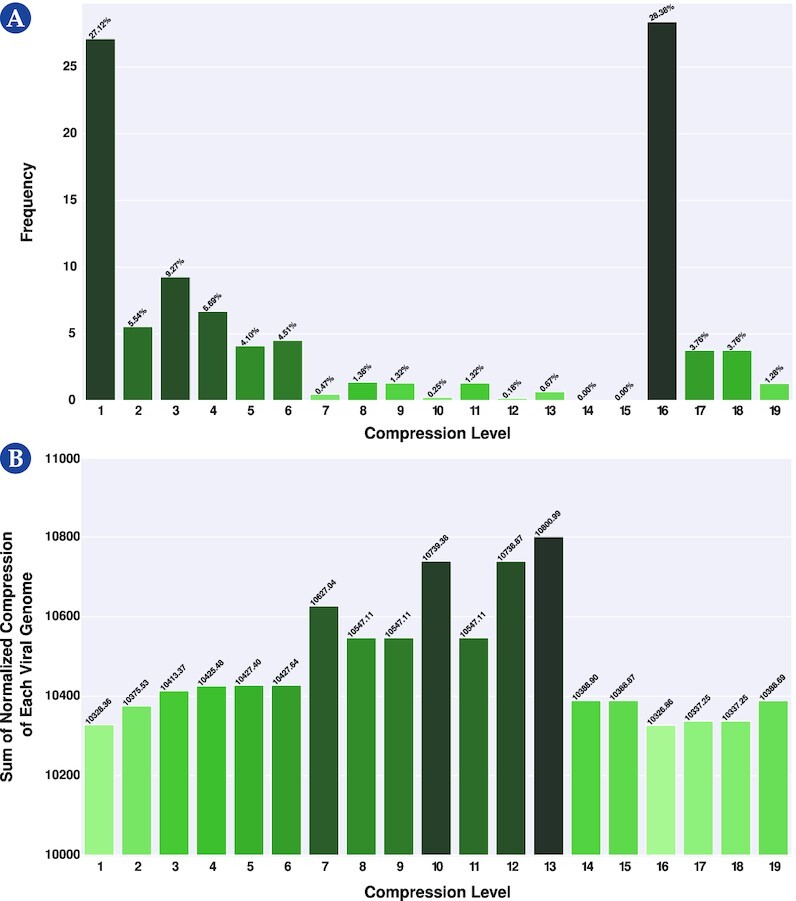
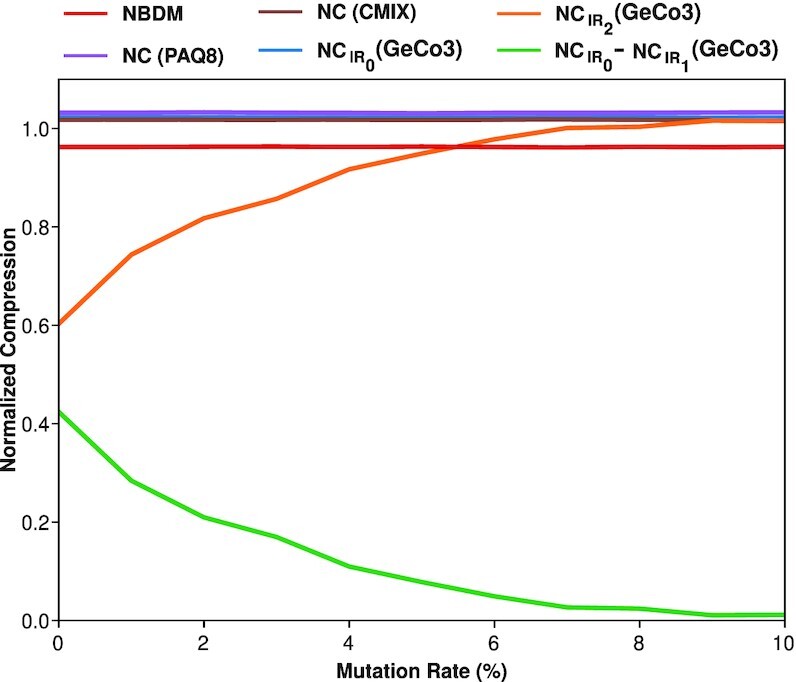
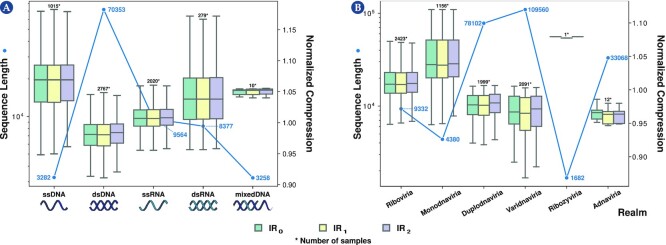
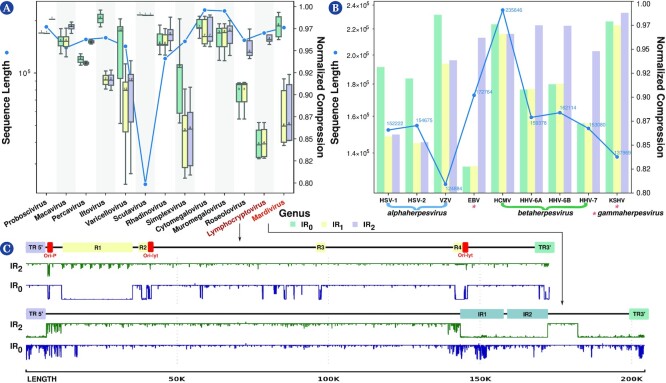
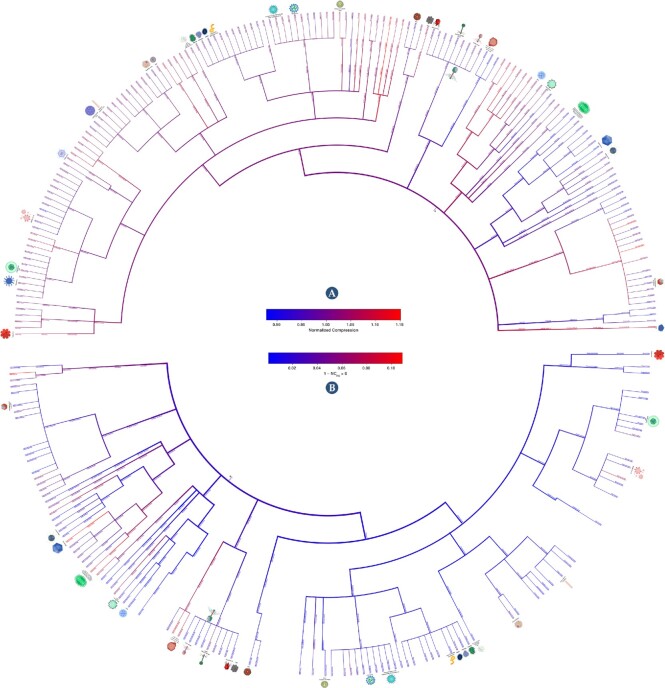
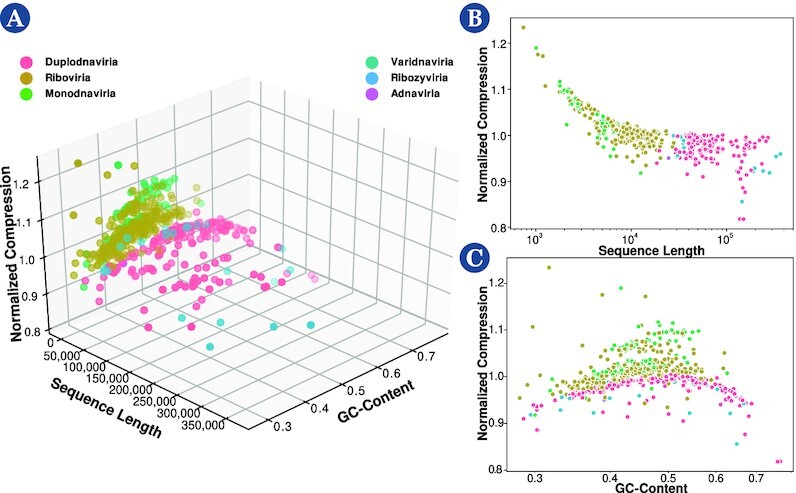
Results: This work provides a comprehensive landscape of the viral genome's complexity (or quantity of information), identifying the most redundant and complex groups regarding their genome sequence while providing their distribution and characteristics at a large and local scale. Moreover, we identify and quantify inverted repeats abundance in viral genomes. For this purpose, we measure the sequence complexity of each available viral genome using data compression, demonstrating that adequate data compressors can efficiently quantify the complexity of viral genome sequences, including subsequences better represented by algorithmic sources (e.g., inverted repeats). Using a state-of-the-art genomic compressor on an extensive viral genomes database, we show that double-stranded DNA viruses are, on average, the most redundant viruses while single-stranded DNA viruses are the least. Contrarily, double-stranded RNA viruses show a lower redundancy relative to single-stranded RNA. Furthermore, we extend the ability of data compressors to quantify local complexity (or information content) in viral genomes using complexity profiles, unprecedently providing a direct complexity analysis of human herpesviruses. We also conceive a features-based classification methodology that can accurately distinguish viral genomes at different taxonomic levels without direct comparisons between sequences. This methodology combines data compression with simple measures such as GC-content percentage and sequence length, followed by machine learning classifiers.
Conclusions: This article presents methodologies and findings that are highly relevant for understanding the patterns of similarity and singularity between viral groups, opening new frontiers for studying viral genomes' organization while depicting the complexity trends and classification components of these genomes at different taxonomic levels. The whole study is supported by an extensive website (https://asilab.github.io/canvas/) for comprehending the viral genome characterization using dynamic and interactive approaches.
Keywords: algorithmic information theory; cladograms; data compression; genomics; sequence analysis; viral classification; viruses.
© The Author(s) 2022. Published by Oxford University Press GigaScience.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
AlcoR: alignment-free simulation, mapping, and visualization of low-complexity regions in biological data.Gigascience. 2022 Dec 28;12:giad101. doi: 10.1093/gigascience/giad101. Epub 2023 Dec 13. Gigascience. 2022. PMID: 38091509 Free PMC article.
-
Toward an Alignment-Free Method for Feature Extraction and Accurate Classification of Viral Sequences.J Comput Biol. 2019 Jun;26(6):519-535. doi: 10.1089/cmb.2018.0239. Epub 2019 May 3. J Comput Biol. 2019. PMID: 31050550
-
A hybrid pipeline for reconstruction and analysis of viral genomes at multi-organ level.Gigascience. 2020 Aug 1;9(8):giaa086. doi: 10.1093/gigascience/giaa086. Gigascience. 2020. PMID: 32815536 Free PMC article.
-
Viral genome sequencing methods: benefits and pitfalls of current approaches.Biochem Soc Trans. 2024 Jun 26;52(3):1431-1447. doi: 10.1042/BST20231322. Biochem Soc Trans. 2024. PMID: 38747720 Free PMC article. Review.
-
Viral Complexity.Biomolecules. 2022 Jul 30;12(8):1061. doi: 10.3390/biom12081061. Biomolecules. 2022. PMID: 36008955 Free PMC article. Review.
Cited by
-
Herpesviruses: overview of systematics, genomic complexity and life cycle.Virol J. 2025 May 22;22(1):155. doi: 10.1186/s12985-025-02779-7. Virol J. 2025. PMID: 40399963 Free PMC article. Review.
-
Hecatomb: an integrated software platform for viral metagenomics.Gigascience. 2024 Jan 2;13:giae020. doi: 10.1093/gigascience/giae020. Gigascience. 2024. PMID: 38832467 Free PMC article.
-
Genomic Insights into Neglected Orthobunyaviruses: Molecular Characterization and Phylogenetic Analysis.Viruses. 2025 Mar 13;17(3):406. doi: 10.3390/v17030406. Viruses. 2025. PMID: 40143333 Free PMC article.
-
Temperature modulates dominance of a superinfecting Arctic virus in its unicellular algal host.ISME J. 2024 Jan 8;18(1):wrae161. doi: 10.1093/ismejo/wrae161. ISME J. 2024. PMID: 39173010 Free PMC article.
References
-
- Edwards RA, Rohwer F. Viral metagenomics. Nat Rev Microbiol. 2005;3(6):504–10. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
