

Fibroblast growth factor receptor 3 overexpression mediates ALK inhibitor resistance in ALK-rearranged non-small cell lung cancer
- PMID: 35950895
- PMCID: PMC9633314
- DOI: 10.1111/cas.15529
Fibroblast growth factor receptor 3 overexpression mediates ALK inhibitor resistance in ALK-rearranged non-small cell lung cancer
Abstract
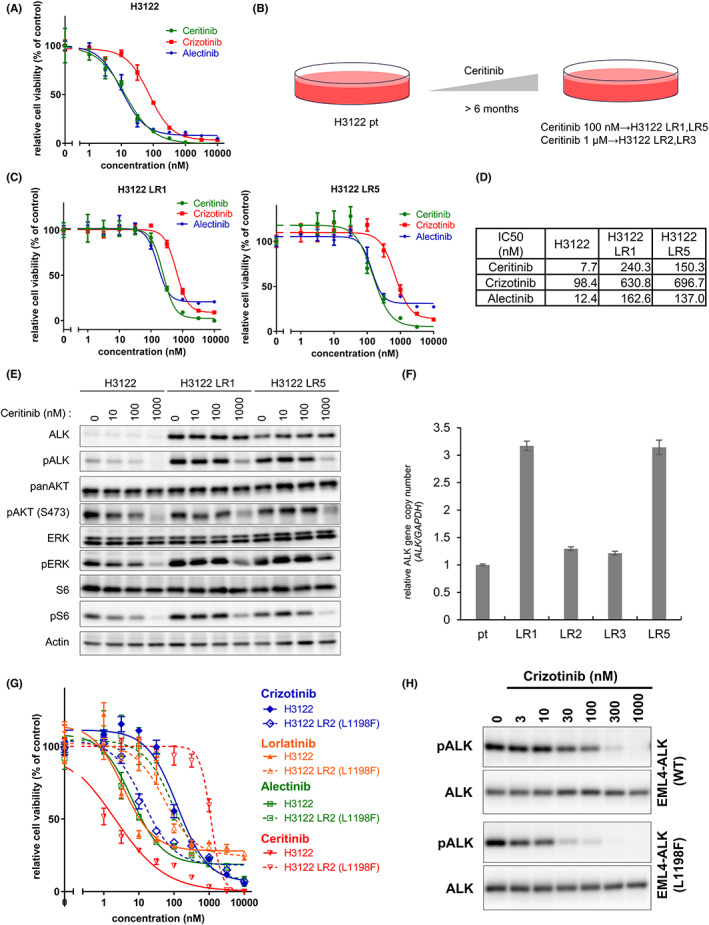
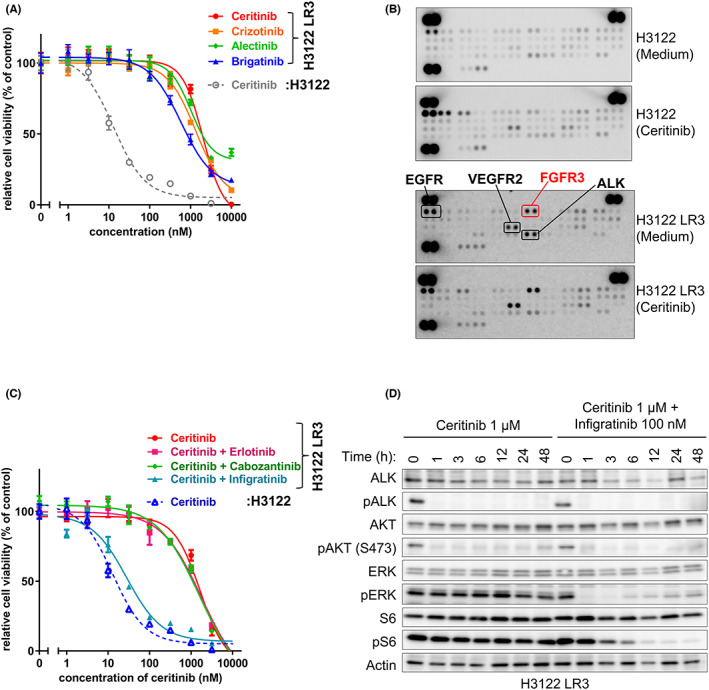
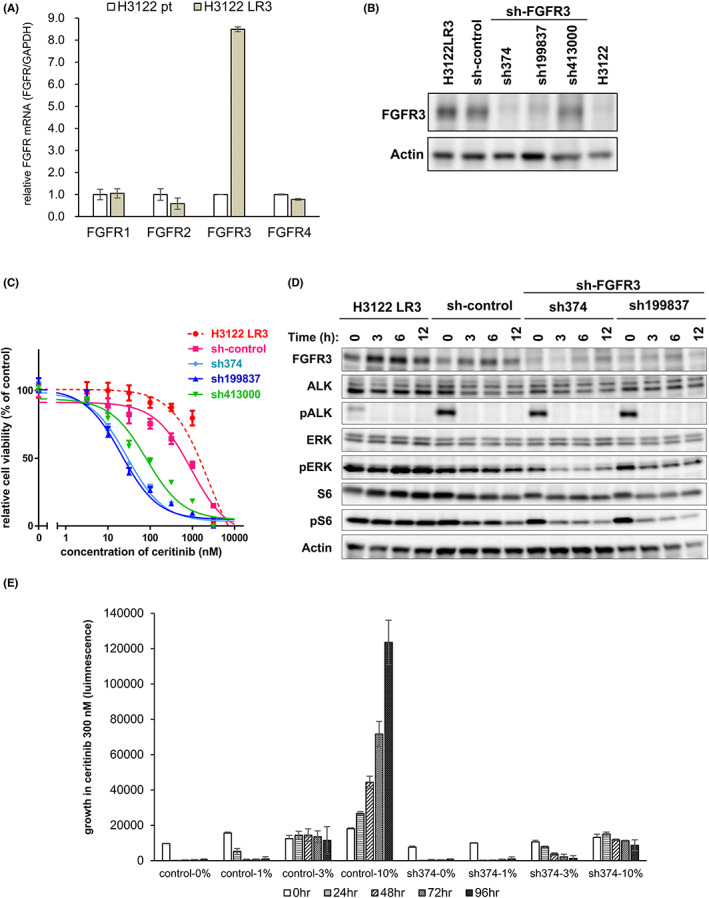
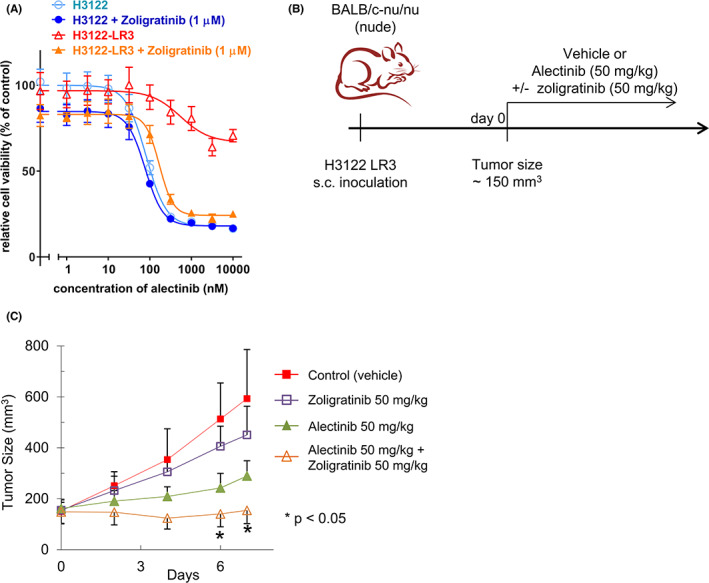
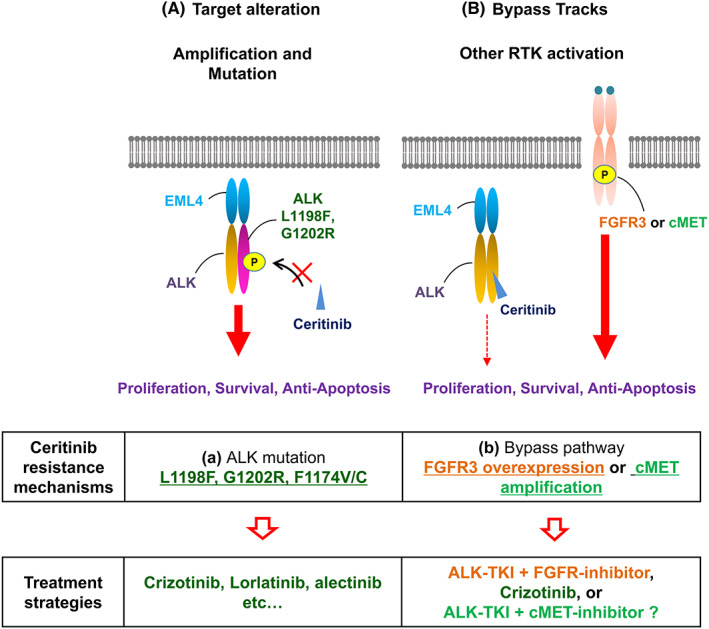
The rearrangement of anaplastic lymphoma kinase (ALK) occurs in 3%-5% of patients with non-small cell lung cancer (NSCLC) and confers sensitivity to ALK-tyrosine kinase inhibitors (TKIs). For the treatment of patients with ALK-rearranged NSCLC, various additional ALK-TKIs have been developed. Ceritinib is a second-generation ALK-TKI and has shown great efficacy in the treatment of patients with both newly diagnosed and crizotinib (a first-generation ALK-TKI)-refractory ALK-rearranged NSCLC. However, tumors can also develop ceritinib resistance. This may result from secondary ALK mutations, but other mechanisms responsible for this have not been fully elucidated. In this study, we explored the mechanisms of ceritinib resistance by establishing ceritinib-resistant, echinoderm microtubule-associated protein-like 4 (EML4)-ALK-positive H3122 cells and ceritinib-resistant patient-derived cells. We identified a mechanism of ceritinib resistance induced by bypass signals that is mediated by the overexpression and activation of fibroblast growth factor receptor 3 (FGFR3). FGFR3 knockdown by small hairpin RNA or treatment with FGFR inhibitors was found to resensitize the resistant cells to ceritinib in vitro and in vivo. FGFR ligands from either human serum or fetal bovine serum were able to activate FGFR3 and induce ceritinib resistance. A detailed analysis of ceritinib-resistant patient-derived specimens confirmed that tyrosine-protein kinase Met (cMET) amplification induces ceritinib resistance. Amplified cMET counteractivated EGFR and/or Her3 and induced ceritinib resistance. These results reveal multiple ceritinib resistance mechanisms and suggest that ceritinib resistance might be overcome by identifying precise resistance mechanisms.
Keywords: ALK kinase; FGFR3; ceritinib; drug resistance; lung cancer.
© 2022 The Authors. Cancer Science published by John Wiley & Sons Australia, Ltd on behalf of Japanese Cancer Association.
Figures
References
-
- Cheng L, Li Y, Zhang SB, Teng XD. Molecular pathology of lung cancer: key to personalized medicine. Zhonghua Bing Li Xue Za Zhi. 2012;41(10):715‐720. doi:10.3760/cma.j.issn.0529‐5807.2012.10.019 - DOI - PubMed
-
- Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4‐ALK fusion gene in non‐small‐cell lung cancer. Nature. 2007;448(7153):561‐566. - PubMed
-
- Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. 2015;21(10):2227‐2235. doi:10.1158/1078‐0432.CCR‐14‐2791 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
