

Heterozygous variants in SIX3 and POU1F1 cause pituitary hormone deficiency in mouse and man
- PMID: 35951005
- PMCID: PMC9851746
- DOI: 10.1093/hmg/ddac192
Heterozygous variants in SIX3 and POU1F1 cause pituitary hormone deficiency in mouse and man
Abstract
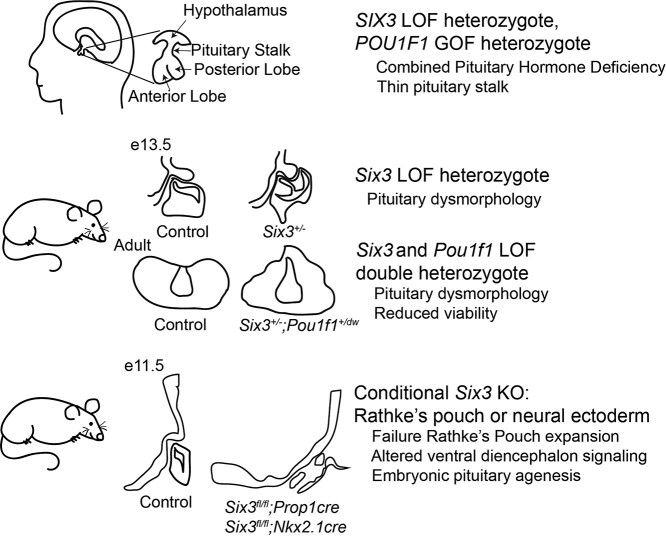
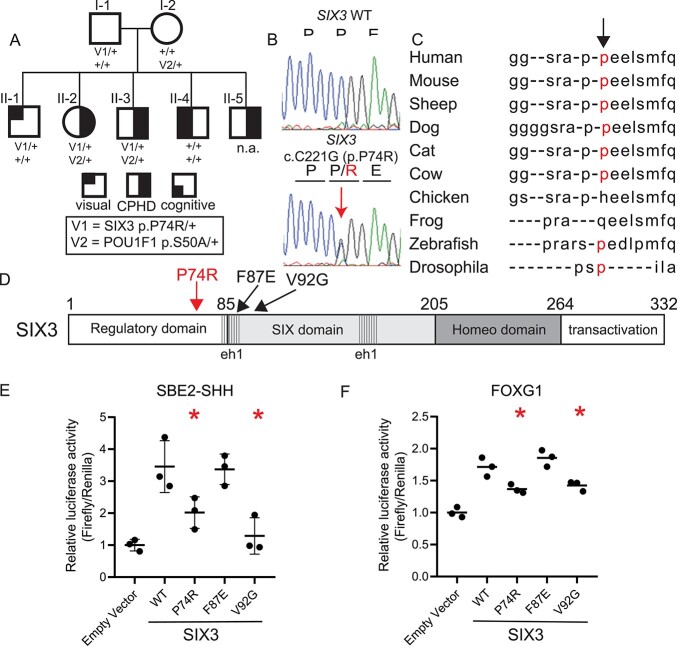
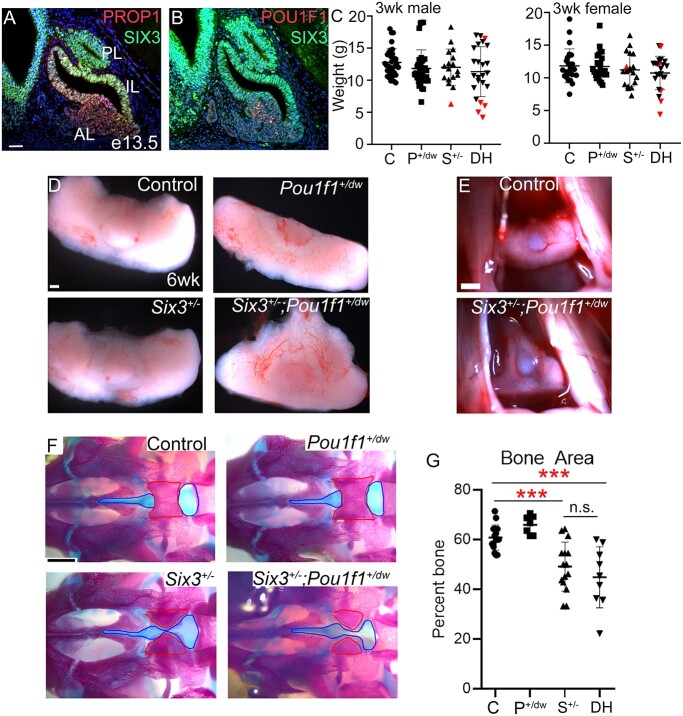
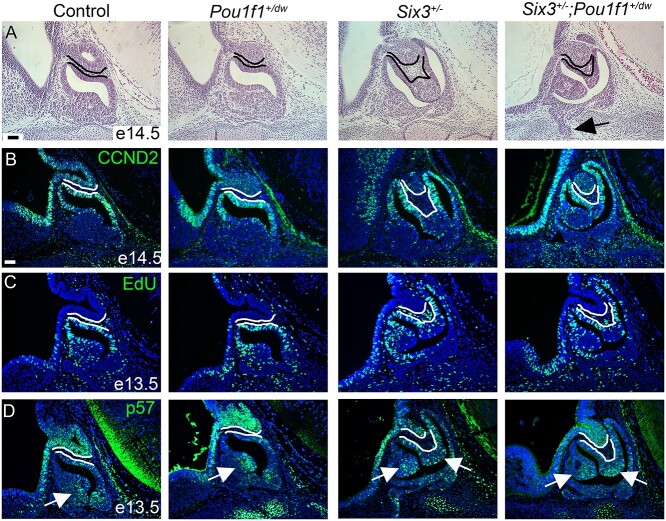
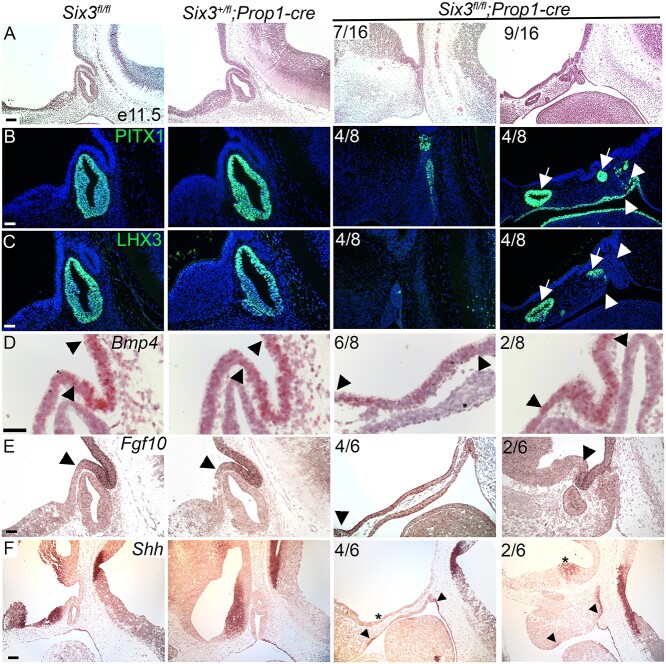
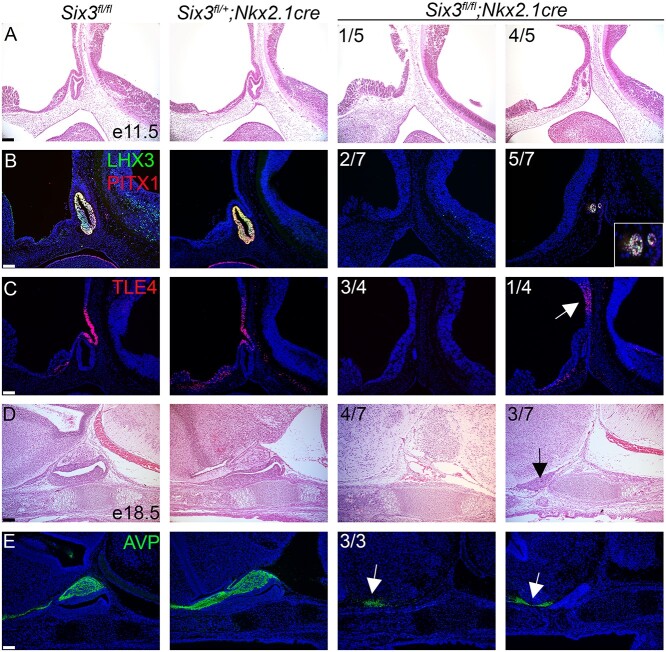
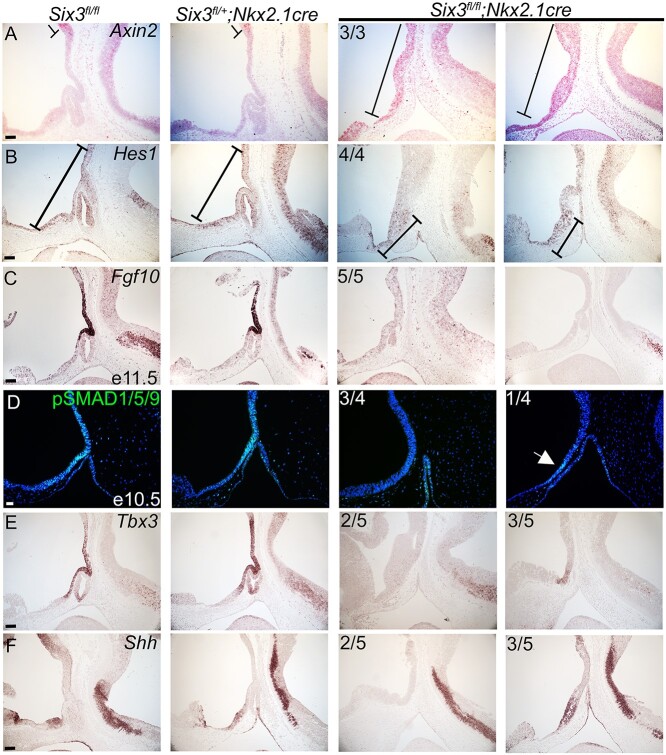
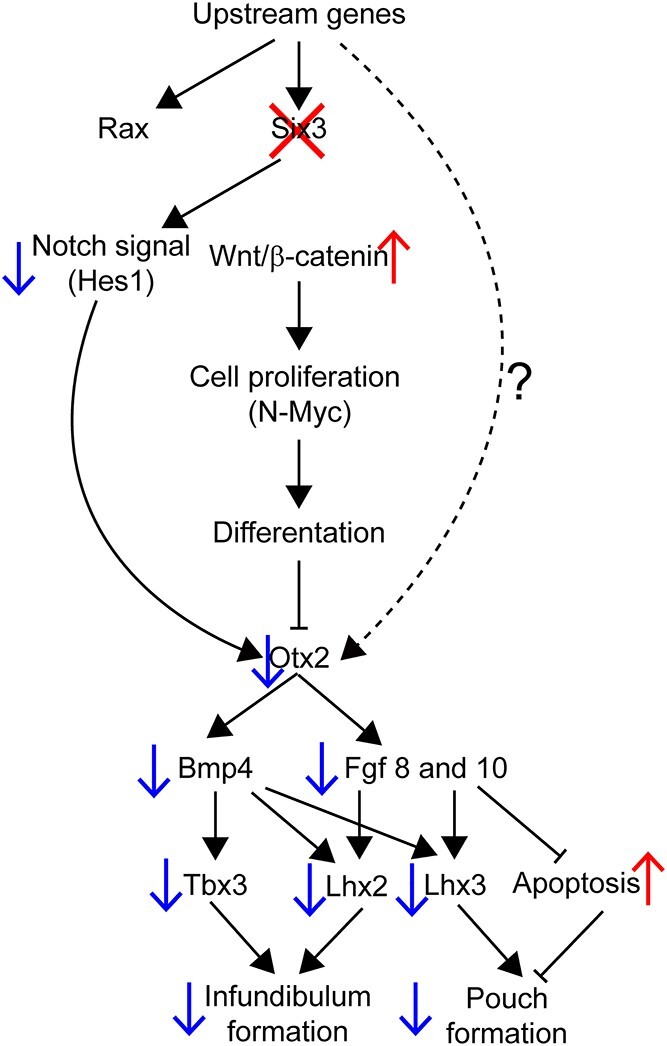
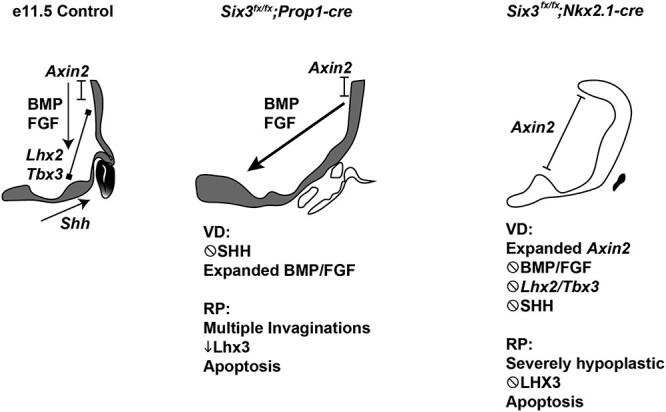
Congenital hypopituitarism is a genetically heterogeneous condition that is part of a spectrum disorder that can include holoprosencephaly. Heterozygous mutations in SIX3 cause variable holoprosencephaly in humans and mice. We identified two children with neonatal hypopituitarism and thin pituitary stalk who were doubly heterozygous for rare, likely deleterious variants in the transcription factors SIX3 and POU1F1. We used genetically engineered mice to understand the disease pathophysiology. Pou1f1 loss-of-function heterozygotes are unaffected; Six3 heterozygotes have pituitary gland dysmorphology and incompletely ossified palate; and the Six3+/-; Pou1f1+/dw double heterozygote mice have a pronounced phenotype, including pituitary growth through the palate. The interaction of Pou1f1 and Six3 in mice supports the possibility of digenic pituitary disease in children. Disruption of Six3 expression in the oral ectoderm completely ablated anterior pituitary development, and deletion of Six3 in the neural ectoderm blocked the development of the pituitary stalk and both anterior and posterior pituitary lobes. Six3 is required in both oral and neural ectodermal tissues for the activation of signaling pathways and transcription factors necessary for pituitary cell fate. These studies clarify the mechanism of SIX3 action in pituitary development and provide support for a digenic basis for hypopituitarism.
© The Author(s) 2022. Published by Oxford University Press.
Figures
References
-
- Budny, B., Zemojtel, T., Kaluzna, M., Gut, P., Niedziela, M., Obara-Moszynska, M., Rabska-Pietrzak, B., Karmelita-Katulska, K., Stajgis, M., Ambroziak, U. et al. (2020) SEMA3A and IGSF10 are novel contributors to combined pituitary hormone deficiency (CPHD). Front Endocrinol (Lausanne), 11, 368. - PMC - PubMed
-
- Jee, Y.H., Gangat, M., Yeliosof, O., Temnycky, A.G., Vanapruks, S., Whalen, P., Gourgari, E., Bleach, C., Yu, C.H., Marshall, I. et al. (2021) Evidence that the etiology of congenital hypopituitarism has a major genetic component but is infrequently monogenic. Front. Genet., 12, 697549. - PMC - PubMed
-
- Calloni, S.F., Cohen, J.S., Meoded, A., Juusola, J., Triulzi, F.M., Huisman, T., Poretti, A. and Fatemi, A. (2017) Compound heterozygous variants in ROBO1 cause a neurodevelopmental disorder with absence of transverse pontine fibers and thinning of the anterior commissure and corpus callosum. Pediatr. Neurol., 70, 70–74. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
