

Developmental and epileptic encephalopathies: from genetic heterogeneity to phenotypic continuum
- PMID: 35951482
- PMCID: PMC9576177
- DOI: 10.1152/physrev.00063.2021
Developmental and epileptic encephalopathies: from genetic heterogeneity to phenotypic continuum
Abstract
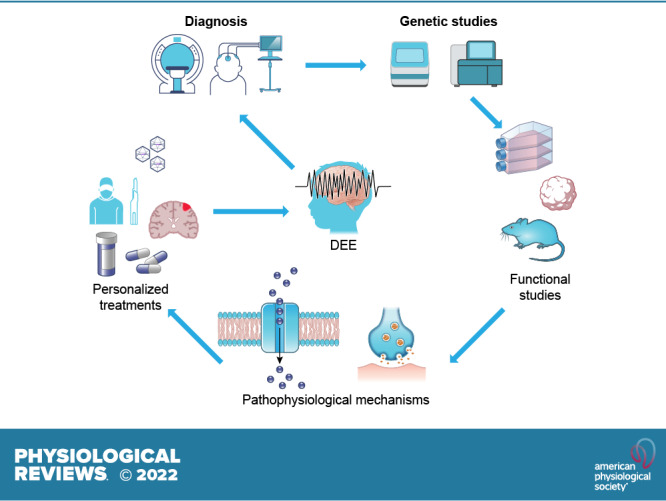
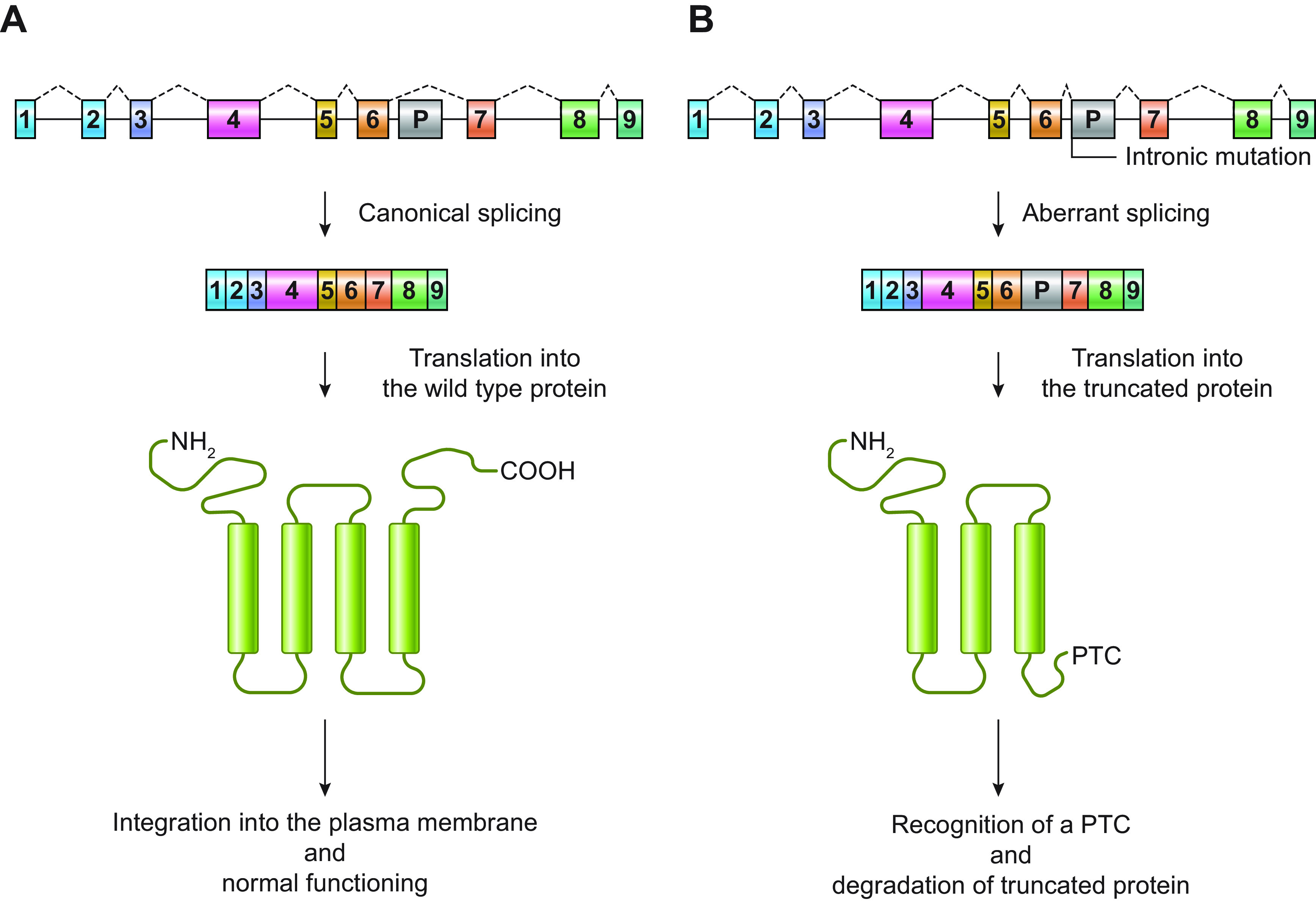
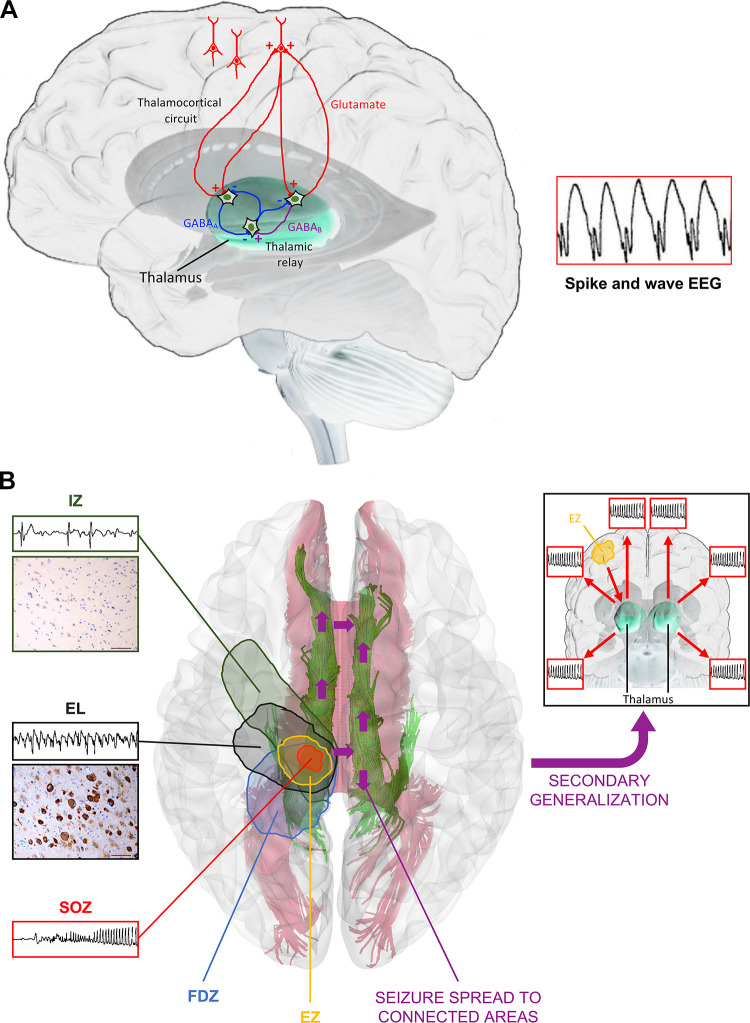
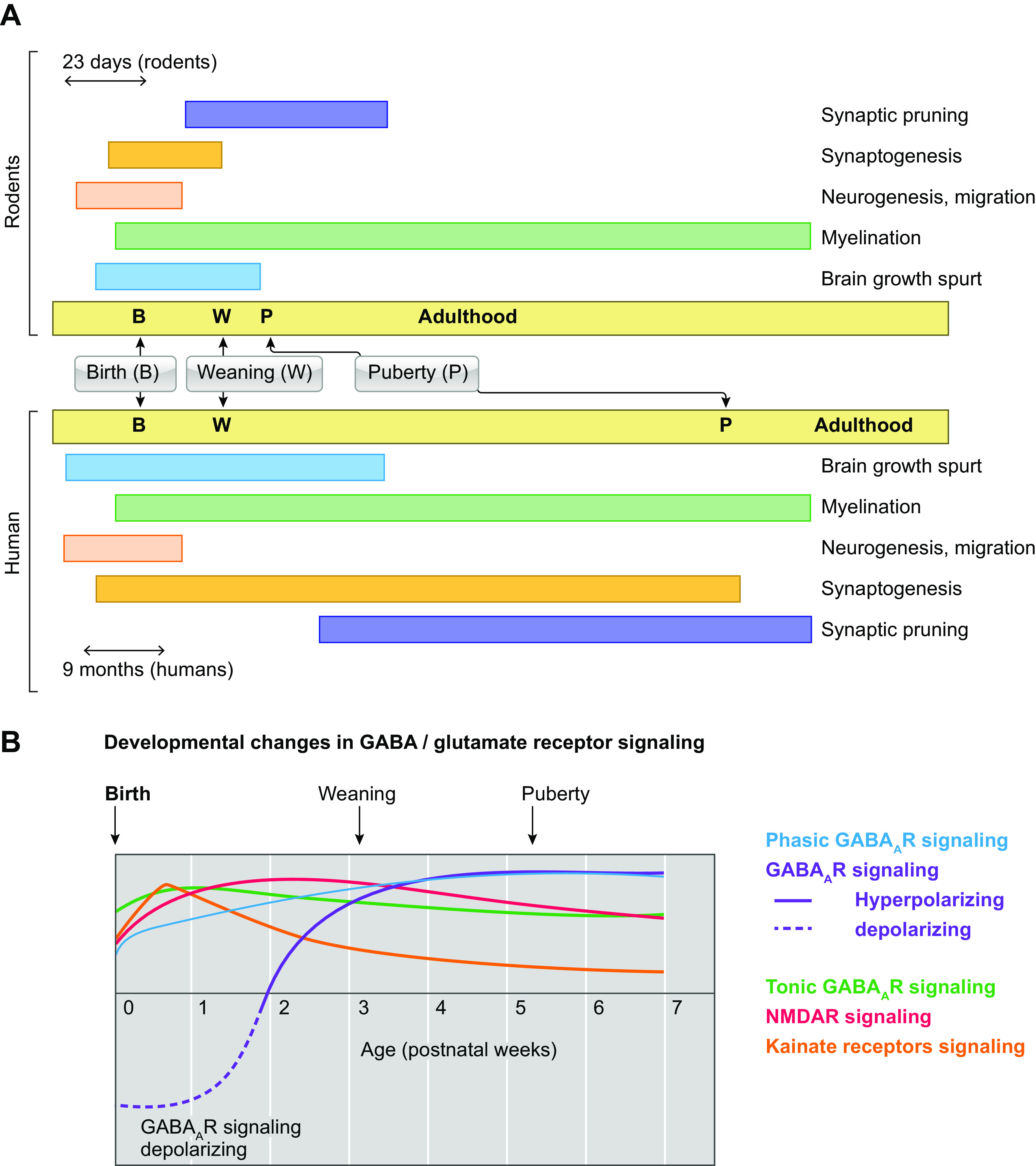
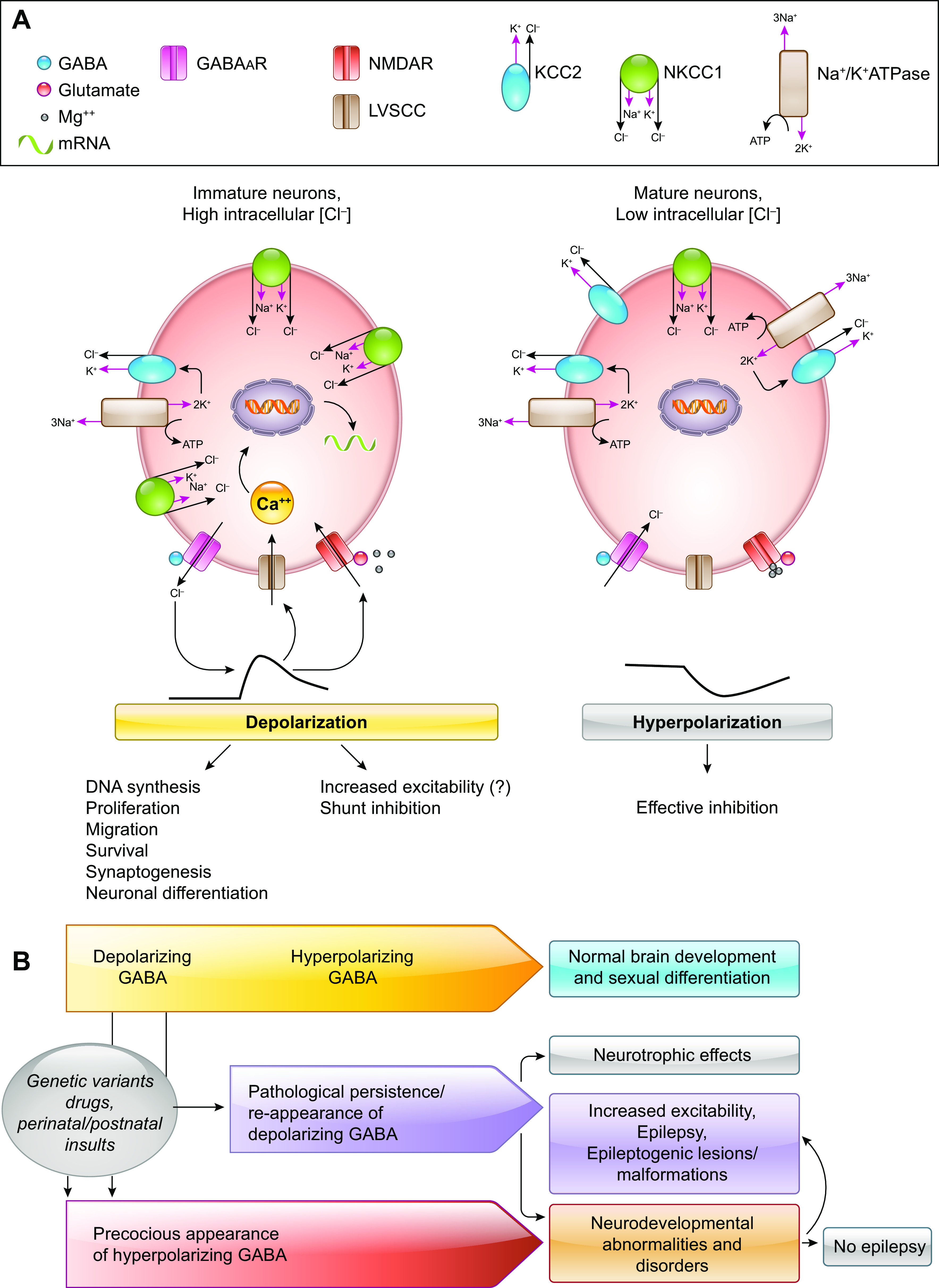
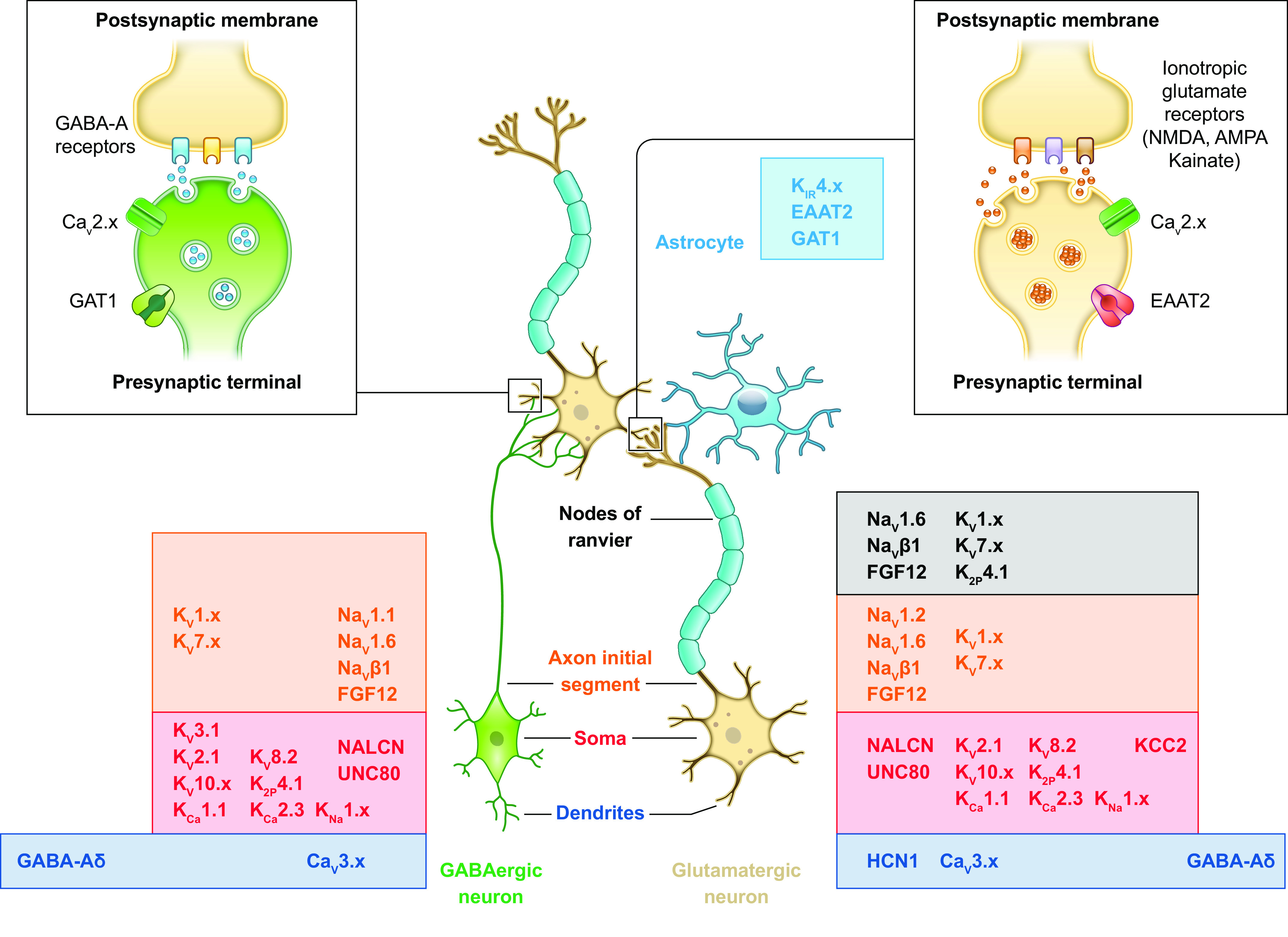
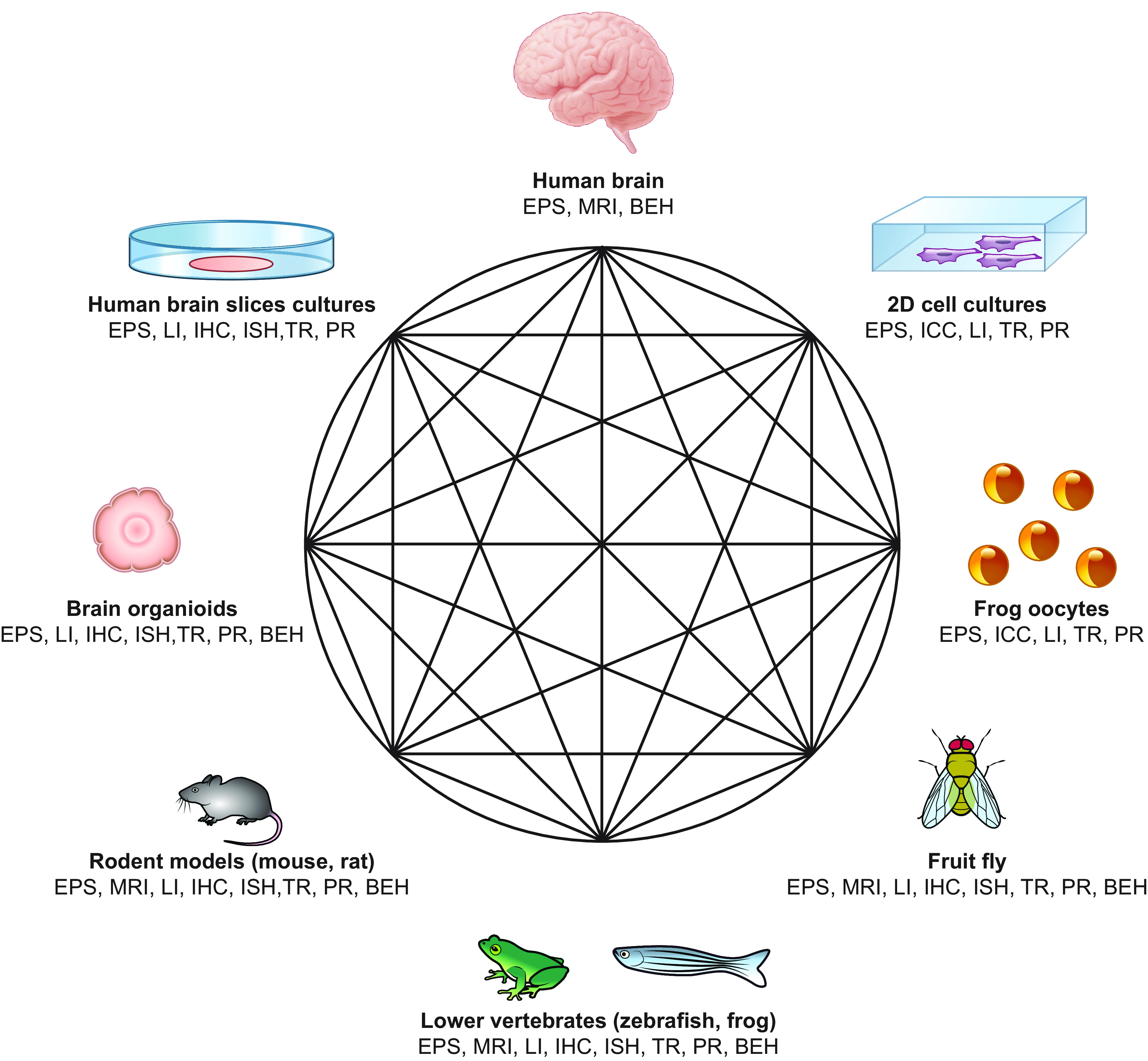
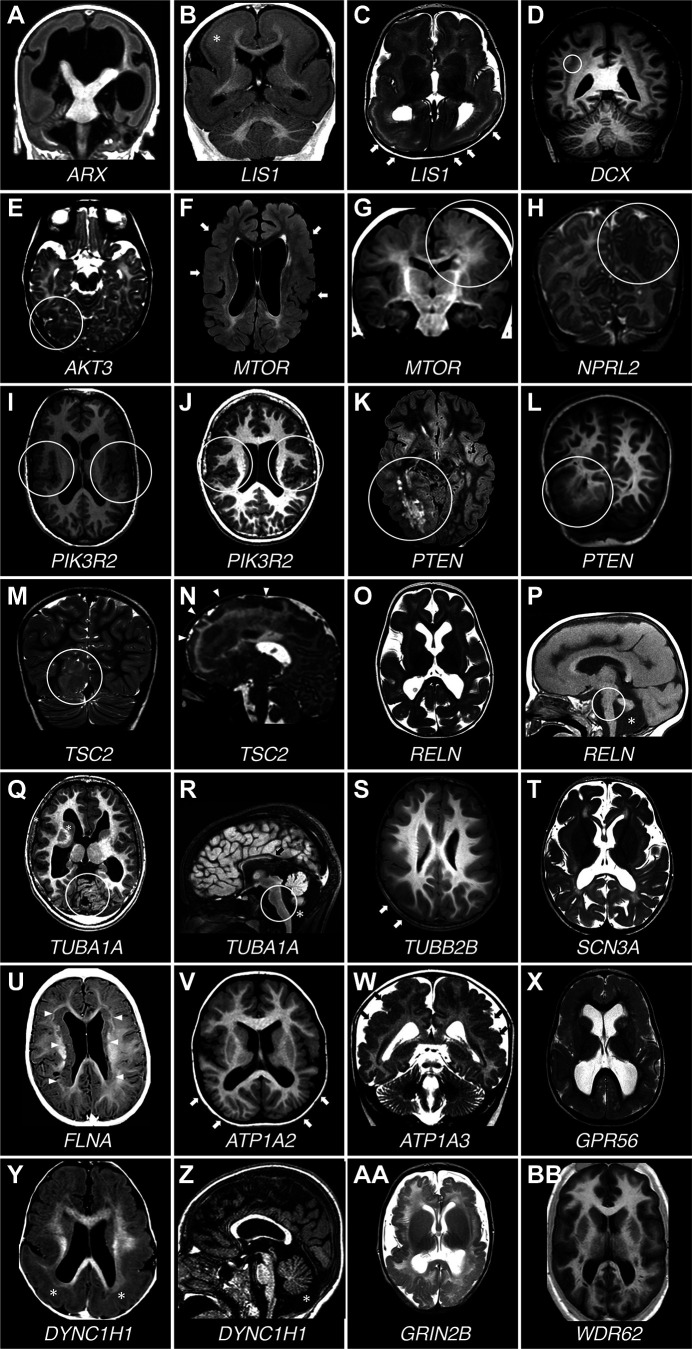
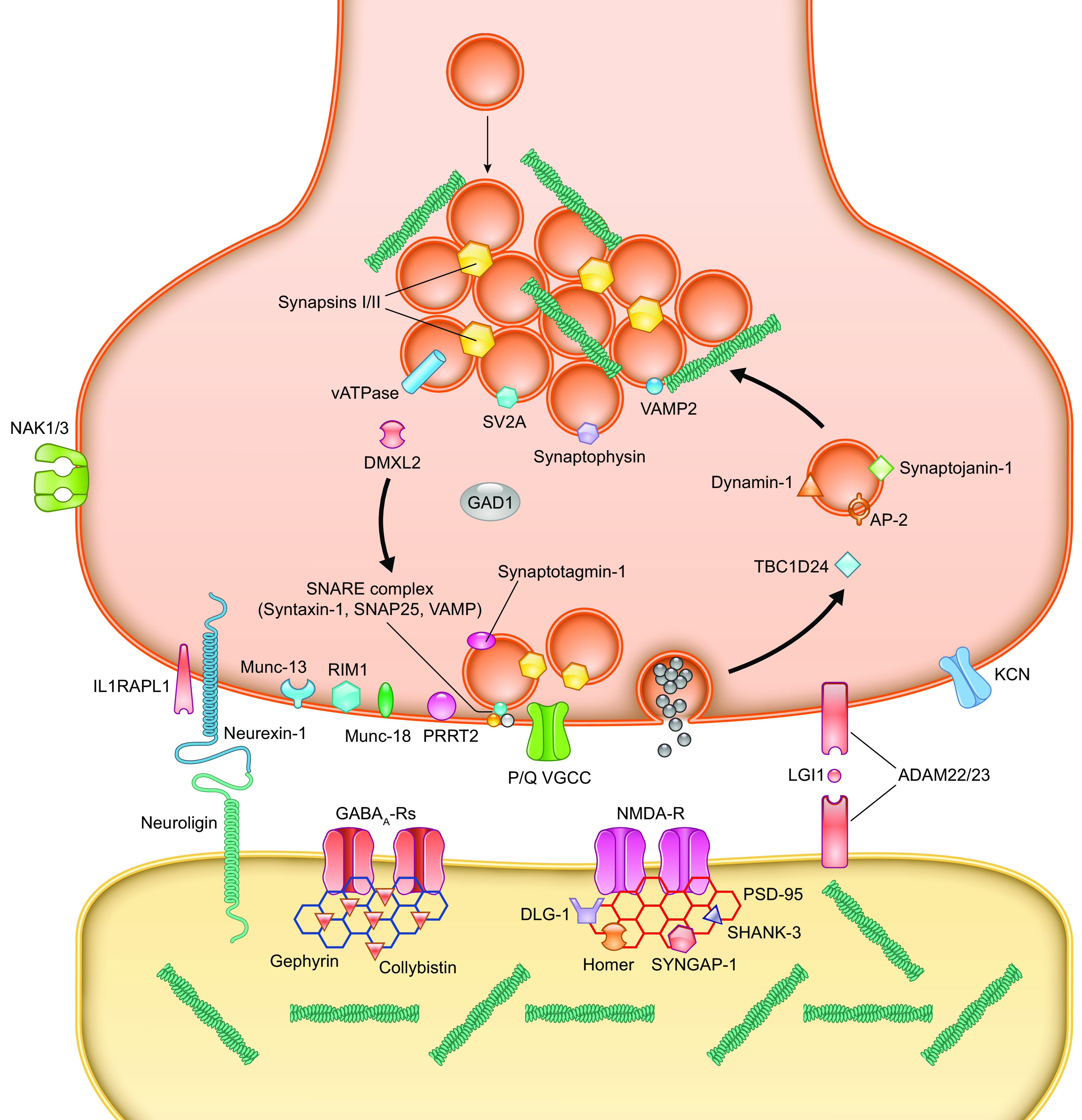
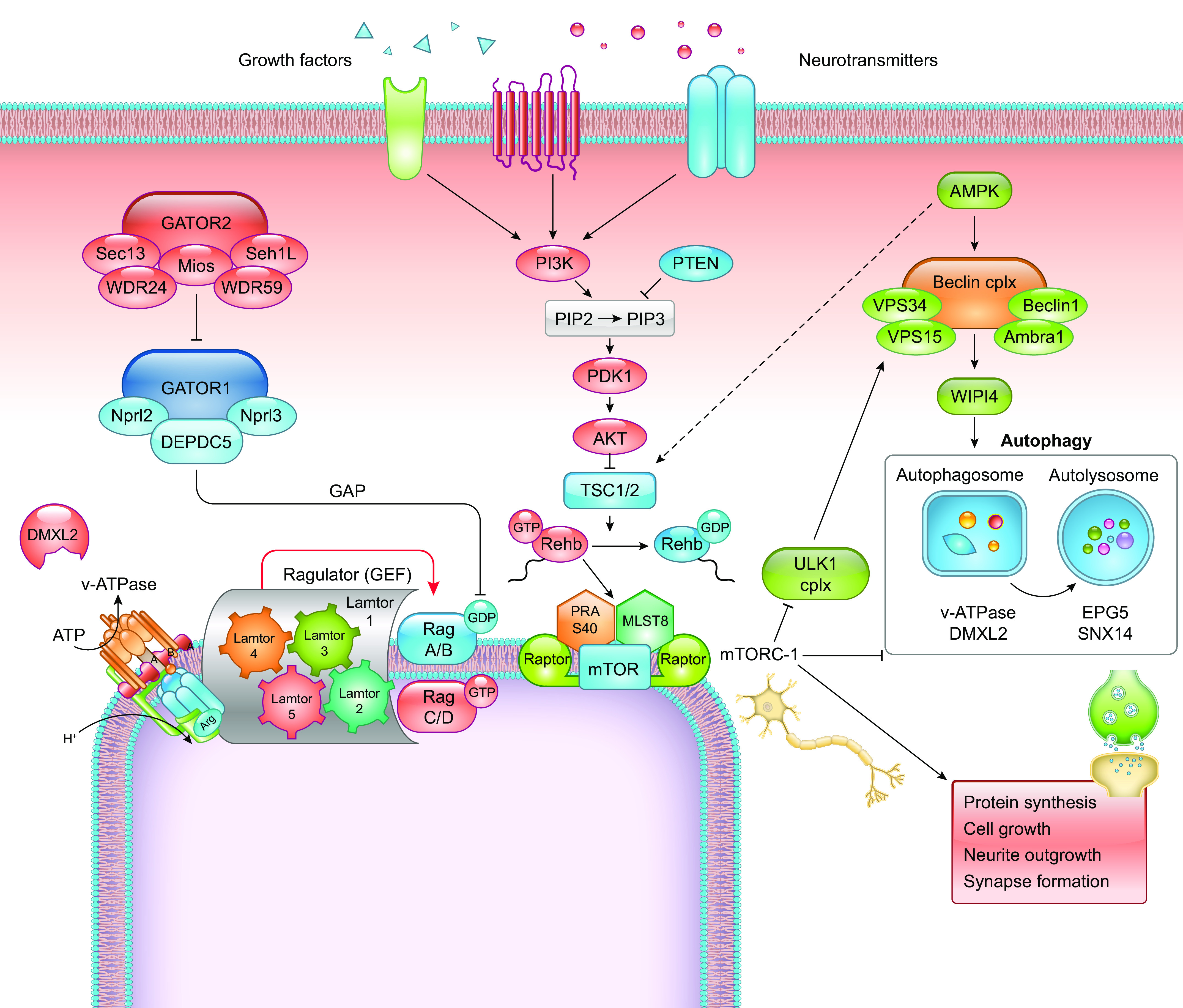
Developmental and epileptic encephalopathies (DEEs) are a heterogeneous group of disorders characterized by early-onset, often severe epileptic seizures and EEG abnormalities on a background of developmental impairment that tends to worsen as a consequence of epilepsy. DEEs may result from both nongenetic and genetic etiologies. Genetic DEEs have been associated with mutations in many genes involved in different functions including cell migration, proliferation, and organization, neuronal excitability, and synapse transmission and plasticity. Functional studies performed in different animal models and clinical trials on patients have contributed to elucidate pathophysiological mechanisms underlying many DEEs and have explored the efficacy of different treatments. Here, we provide an extensive review of the phenotypic spectrum included in the DEEs and of the genetic determinants and pathophysiological mechanisms underlying these conditions. We also provide a brief overview of the most effective treatment now available and of the emerging therapeutic approaches.
Keywords: channelopathies; developmental and epileptic encephalopathies; epileptogenesis; personalized treatment approaches; synaptopathies.
Conflict of interest statement
A.S. Galanopoulou is the Editor-in-Chief of
Figures
References
-
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58: 512–521, 2017. doi: 10.1111/epi.13709. - DOI - PMC - PubMed
-
- Riney K, Bogacz A, Somerville E, Hirsch E, Nabbout R, Scheffer IE, et al. ILAE Classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions (Online). 2021. http://files/1501/Riney_VariableAges_7Apr21.pdf. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical