

Pandemic-scale phylogenomics reveals the SARS-CoV-2 recombination landscape
- PMID: 35952714
- PMCID: PMC9519458
- DOI: 10.1038/s41586-022-05189-9
Pandemic-scale phylogenomics reveals the SARS-CoV-2 recombination landscape
Abstract
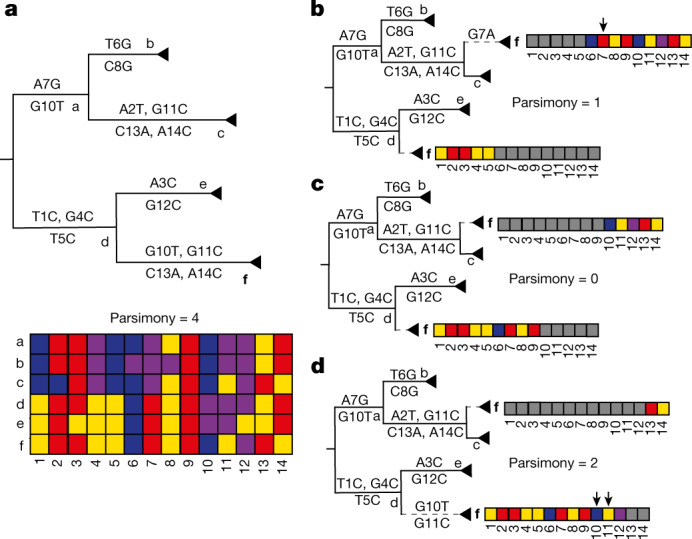
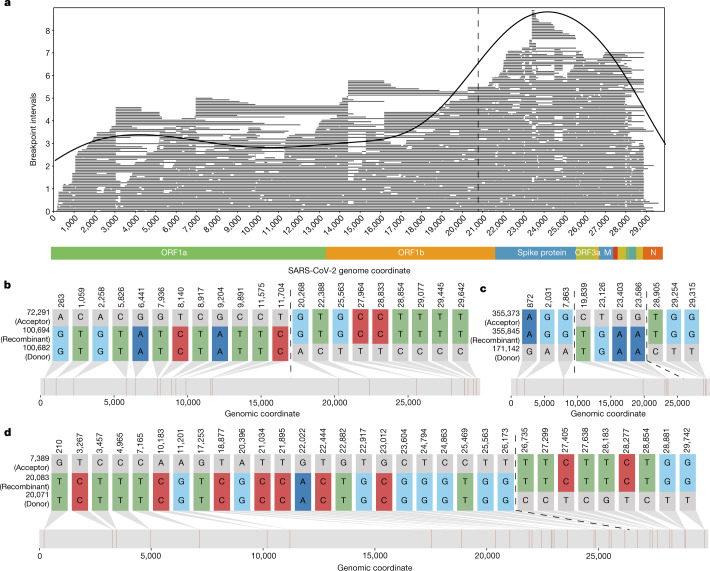
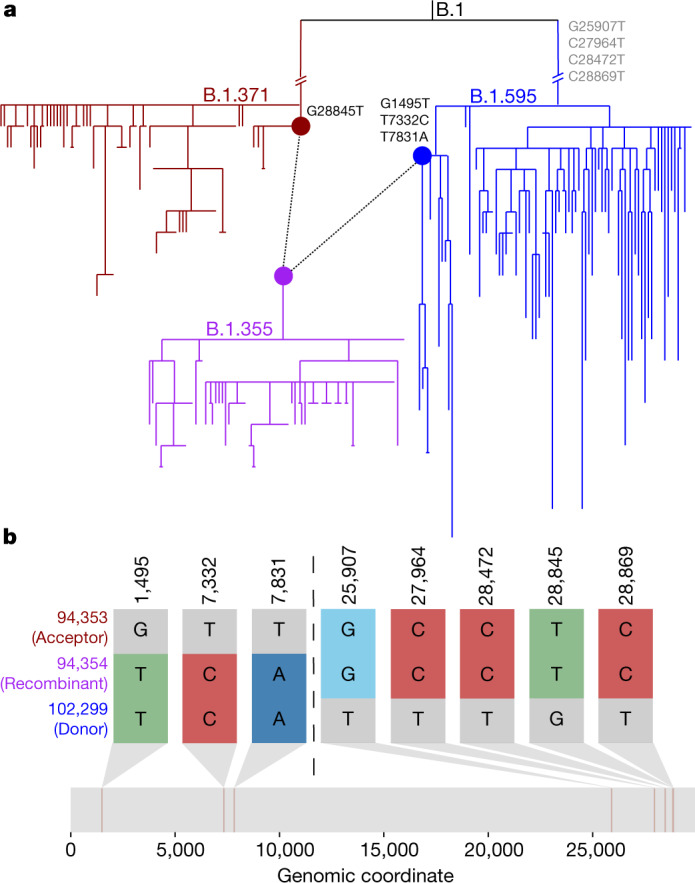
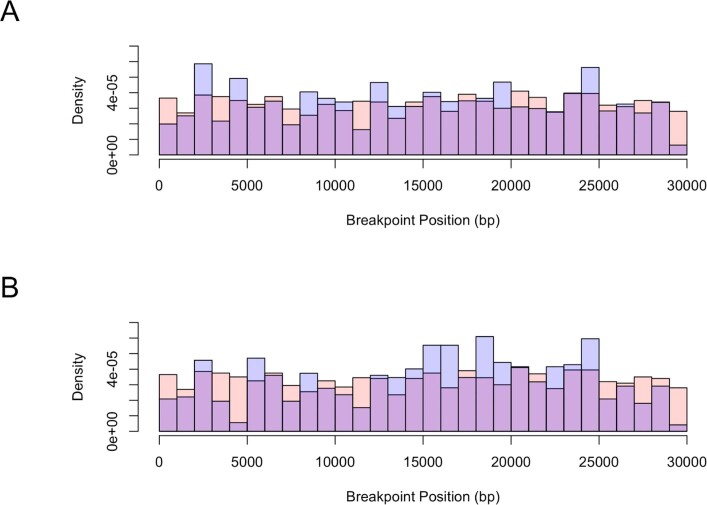
Accurate and timely detection of recombinant lineages is crucial for interpreting genetic variation, reconstructing epidemic spread, identifying selection and variants of interest, and accurately performing phylogenetic analyses1-4. During the SARS-CoV-2 pandemic, genomic data generation has exceeded the capacities of existing analysis platforms, thereby crippling real-time analysis of viral evolution5. Here, we use a new phylogenomic method to search a nearly comprehensive SARS-CoV-2 phylogeny for recombinant lineages. In a 1.6 million sample tree from May 2021, we identify 589 recombination events, which indicate that around 2.7% of sequenced SARS-CoV-2 genomes have detectable recombinant ancestry. Recombination breakpoints are inferred to occur disproportionately in the 3' portion of the genome that contains the spike protein. Our results highlight the need for timely analyses of recombination for pinpointing the emergence of recombinant lineages with the potential to increase transmissibility or virulence of the virus. We anticipate that this approach will empower comprehensive real-time tracking of viral recombination during the SARS-CoV-2 pandemic and beyond.
© 2022. The Author(s).
Conflict of interest statement
R.L. works as an advisor to GISAID. The remaining authors declare no competing interests.
Figures
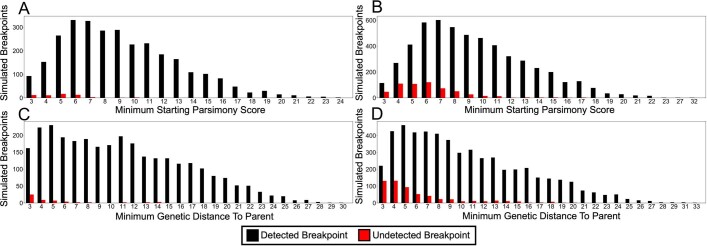

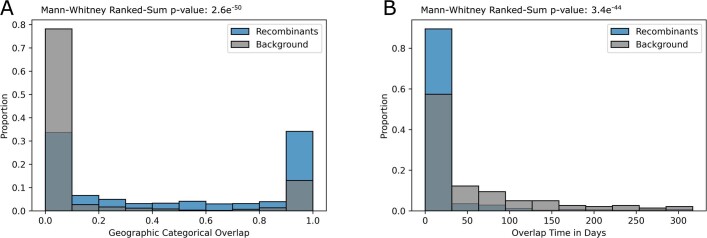
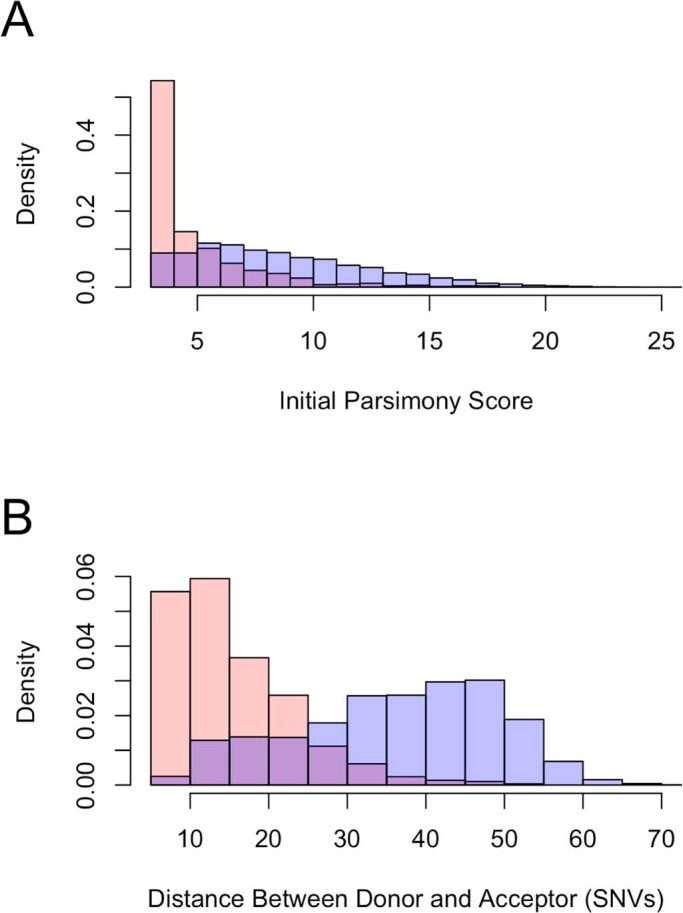
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
