

A multi-tissue atlas of regulatory variants in cattle
- PMID: 35953587
- PMCID: PMC7613894
- DOI: 10.1038/s41588-022-01153-5
A multi-tissue atlas of regulatory variants in cattle
Abstract
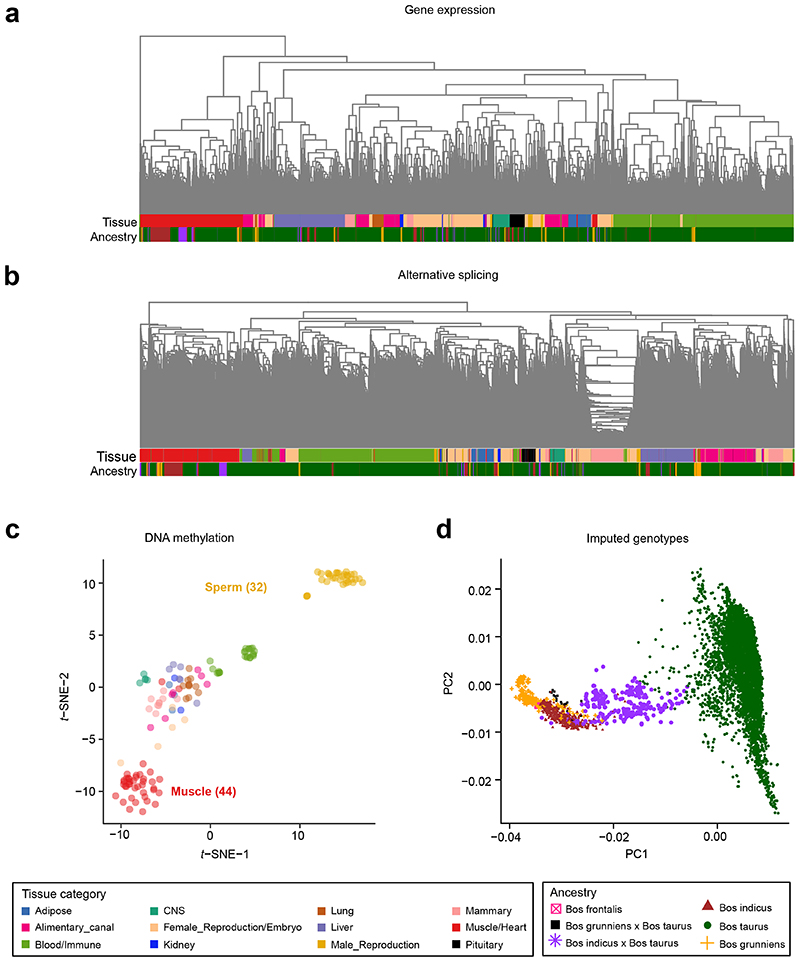
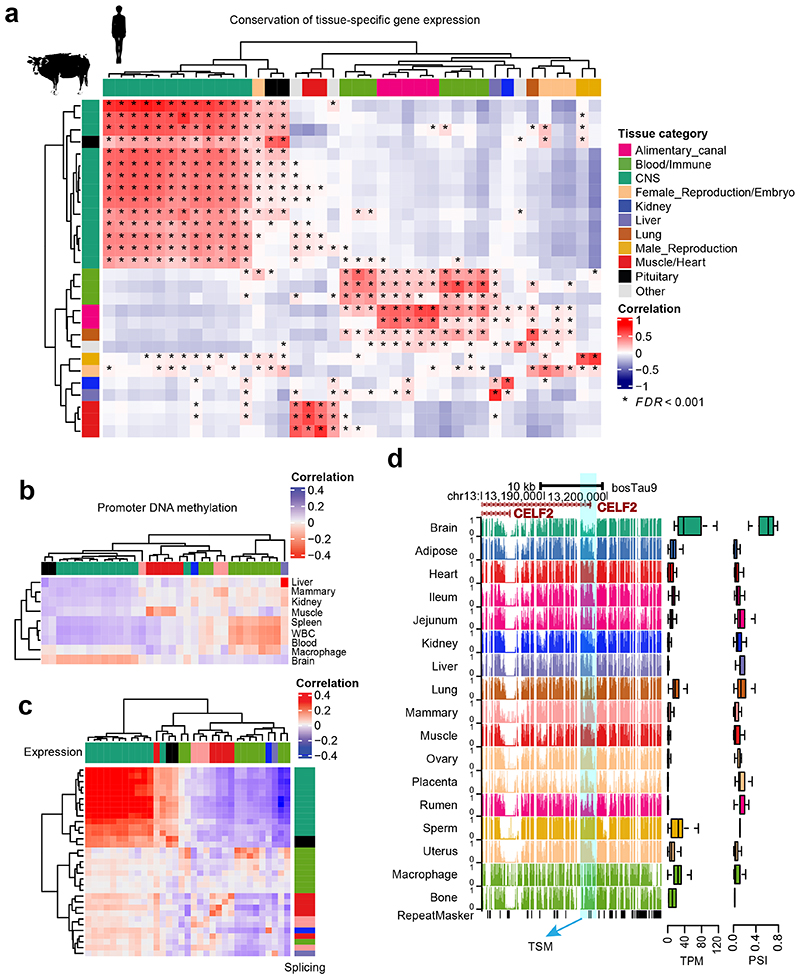
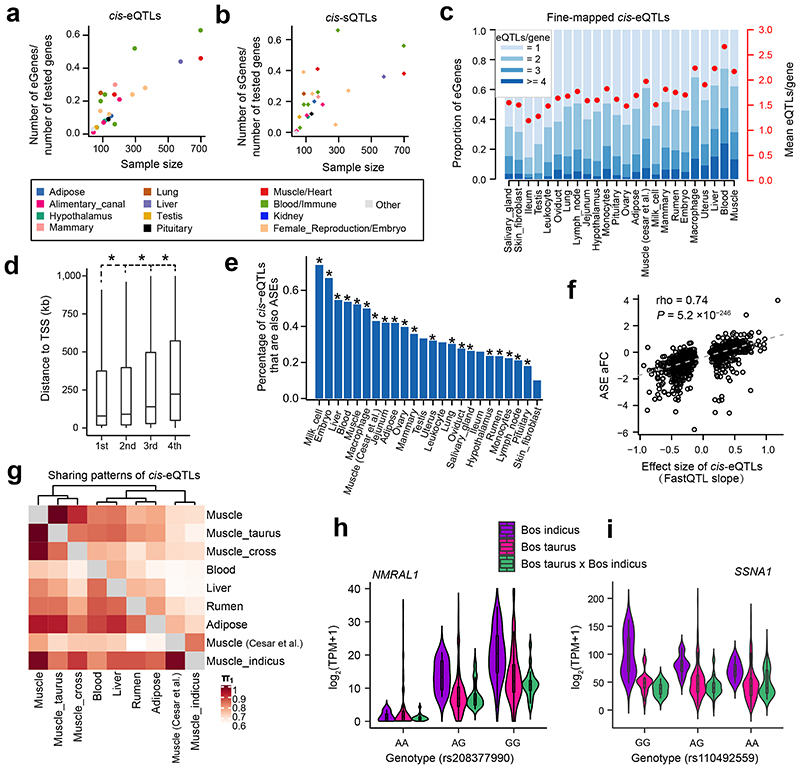
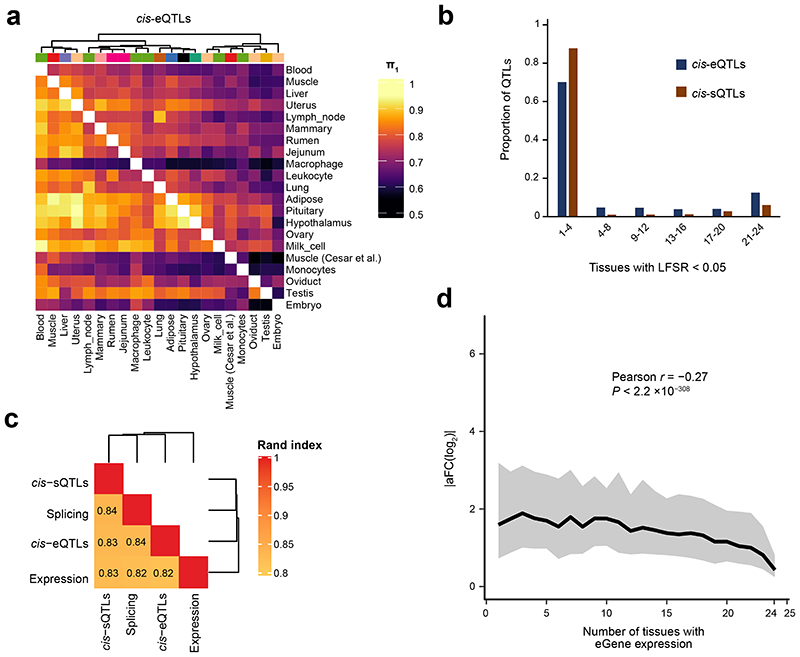
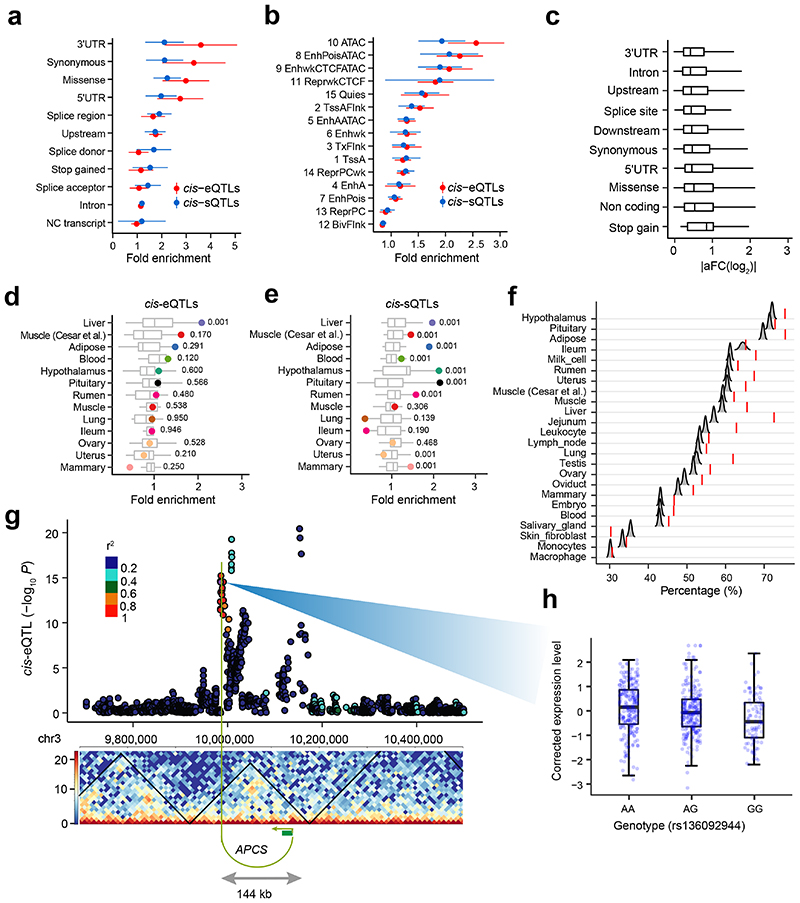
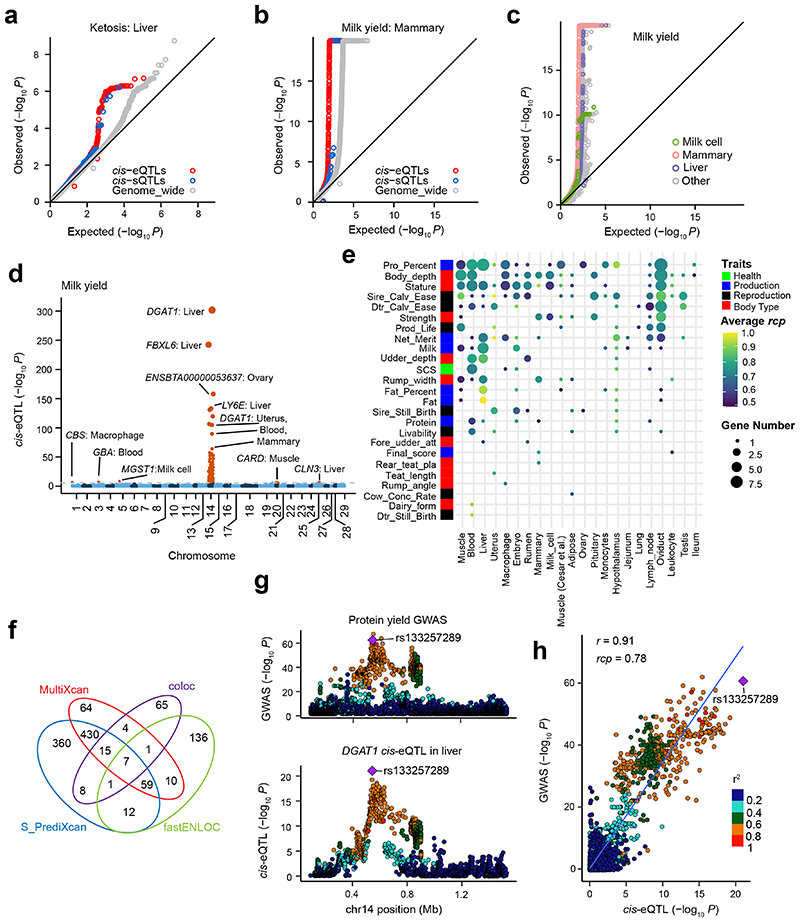
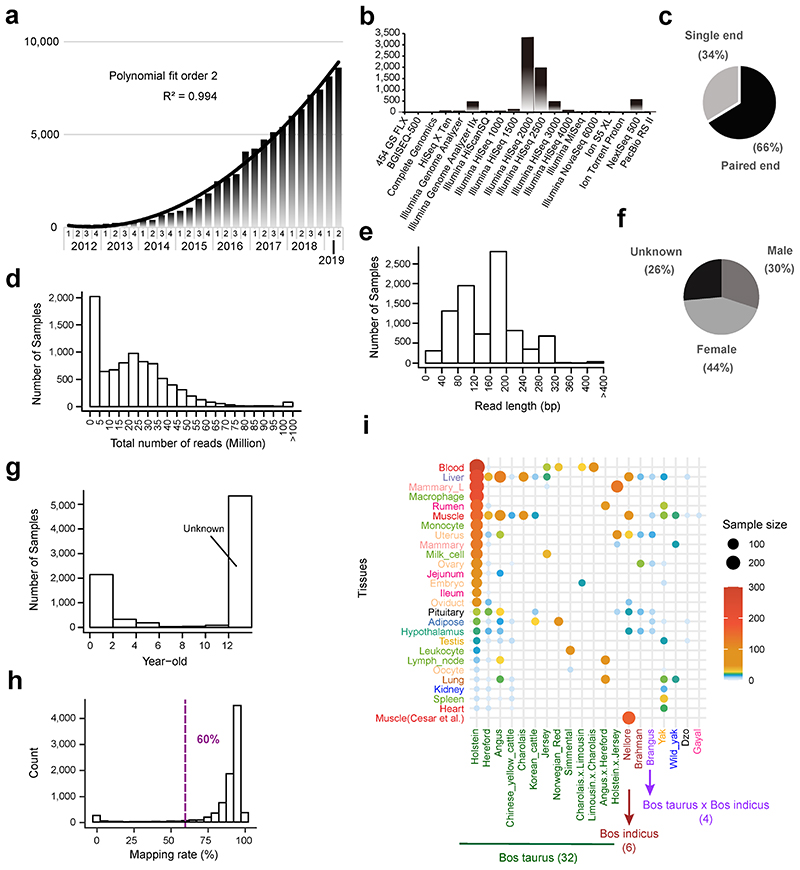
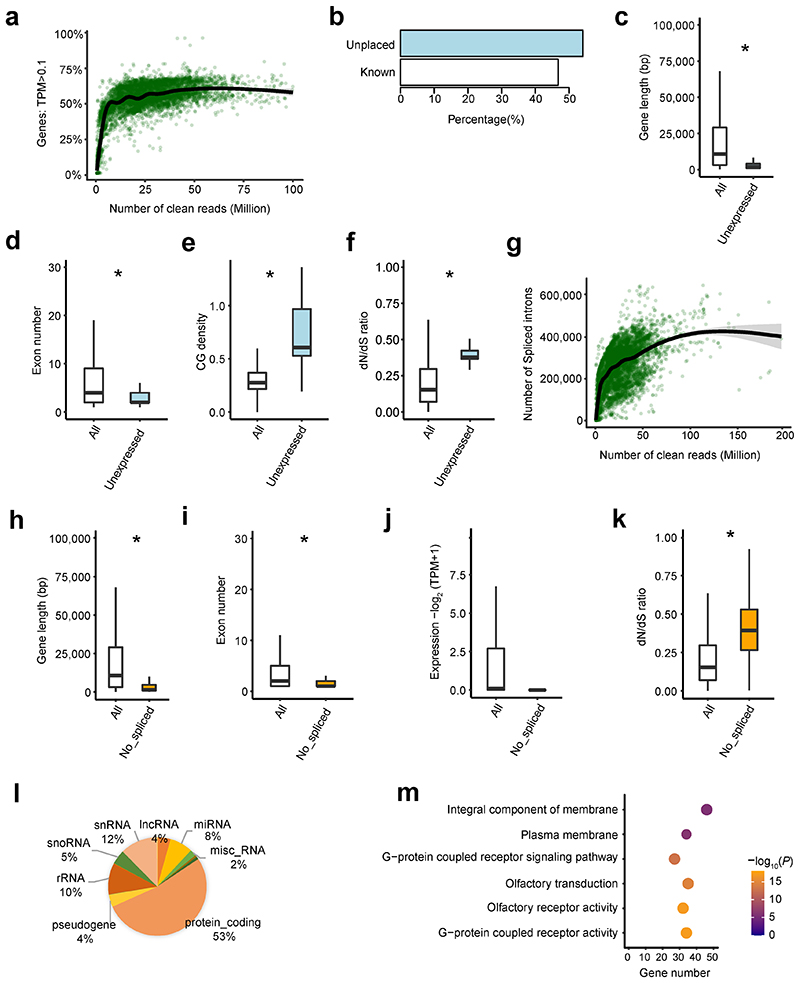
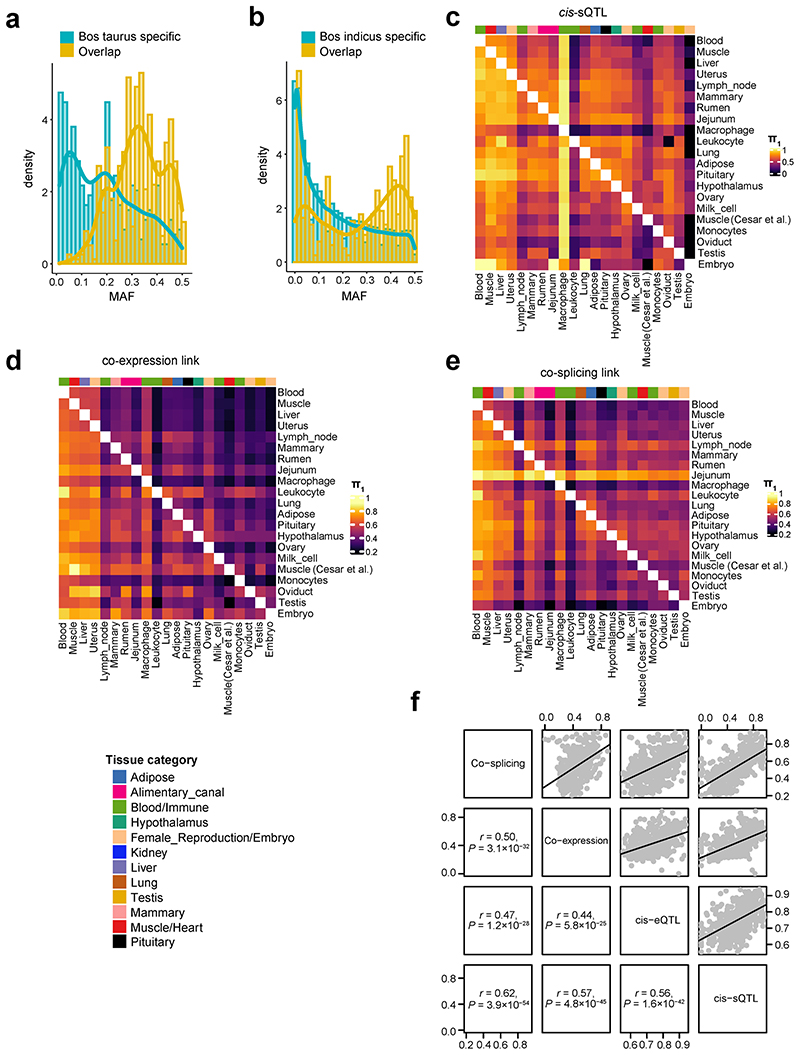
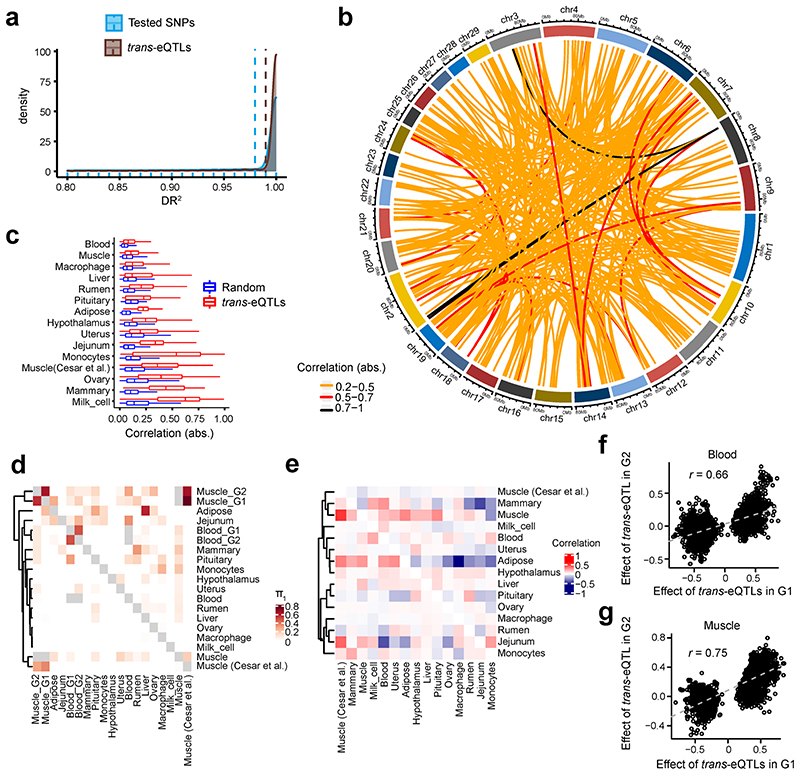
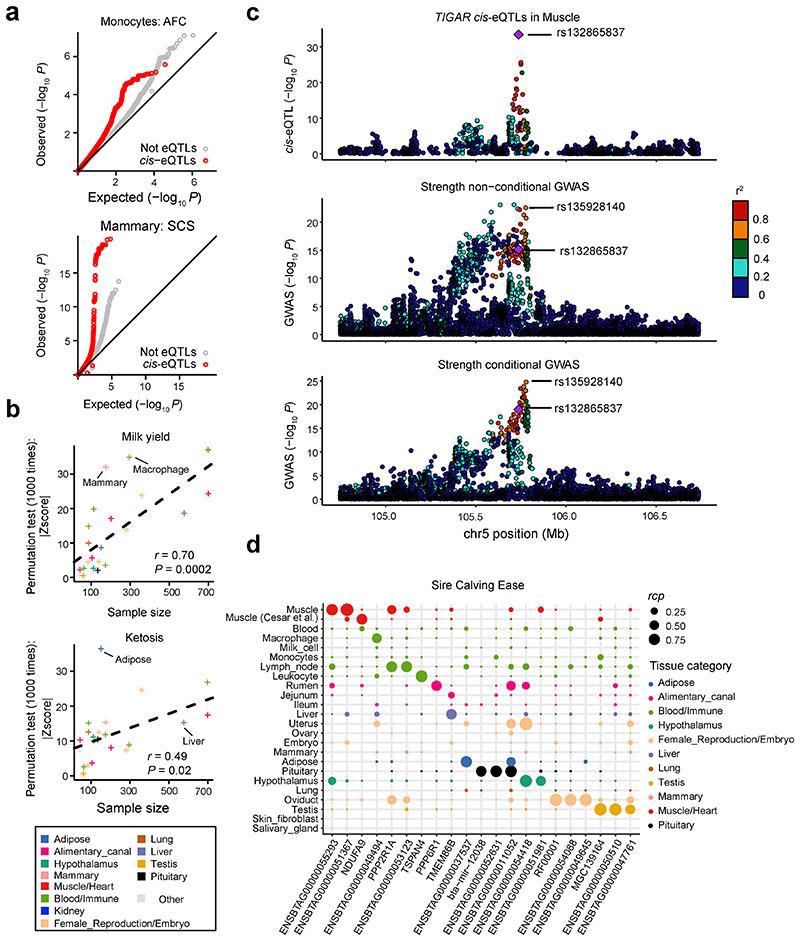
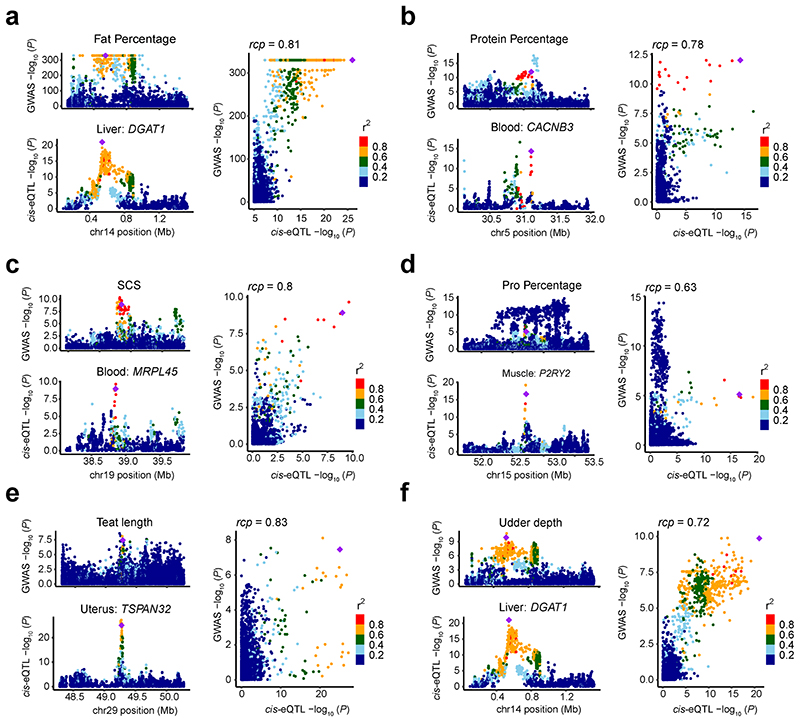
Characterization of genetic regulatory variants acting on livestock gene expression is essential for interpreting the molecular mechanisms underlying traits of economic value and for increasing the rate of genetic gain through artificial selection. Here we build a Cattle Genotype-Tissue Expression atlas (CattleGTEx) as part of the pilot phase of the Farm animal GTEx (FarmGTEx) project for the research community based on 7,180 publicly available RNA-sequencing (RNA-seq) samples. We describe the transcriptomic landscape of more than 100 tissues/cell types and report hundreds of thousands of genetic associations with gene expression and alternative splicing for 23 distinct tissues. We evaluate the tissue-sharing patterns of these genetic regulatory effects, and functionally annotate them using multiomics data. Finally, we link gene expression in different tissues to 43 economically important traits using both transcriptome-wide association and colocalization analyses to decipher the molecular regulatory mechanisms underpinning such agronomic traits in cattle.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
