

MATE1 Deficiency Exacerbates Dofetilide-Induced Proarrhythmia
- PMID: 35955741
- PMCID: PMC9369325
- DOI: 10.3390/ijms23158607
MATE1 Deficiency Exacerbates Dofetilide-Induced Proarrhythmia
Abstract
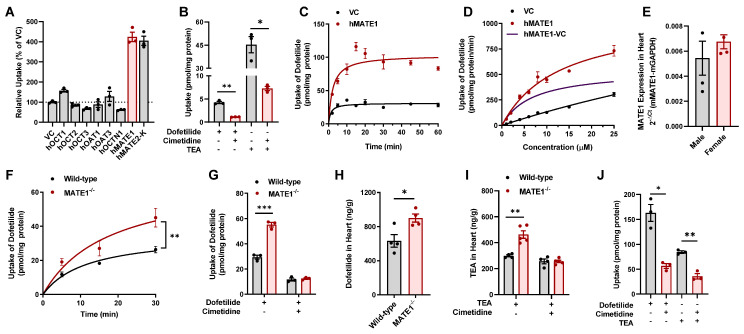
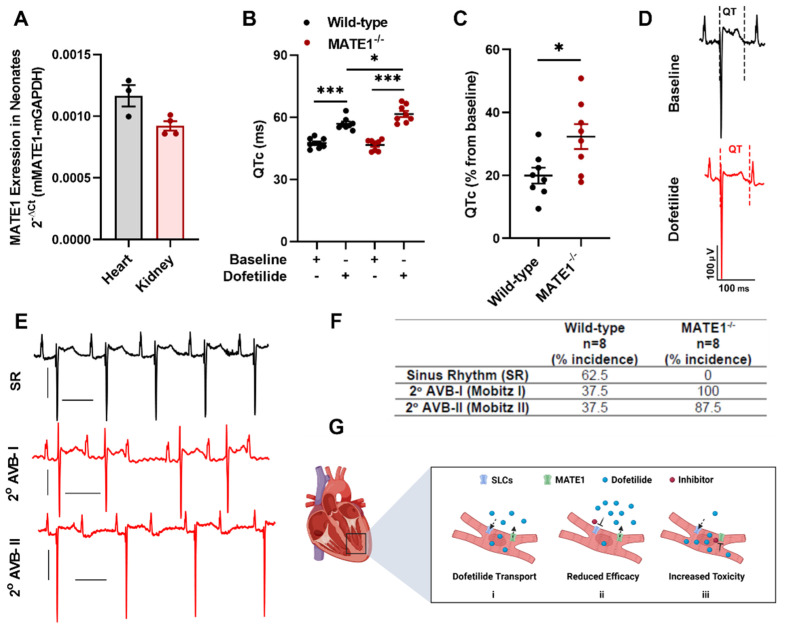
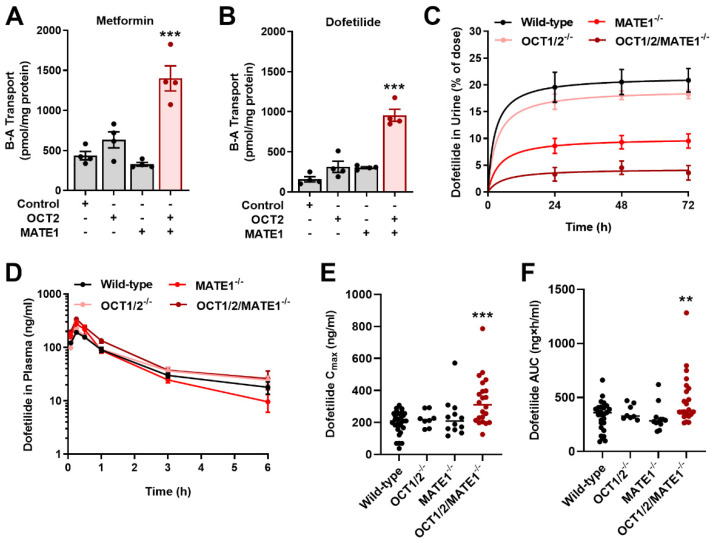
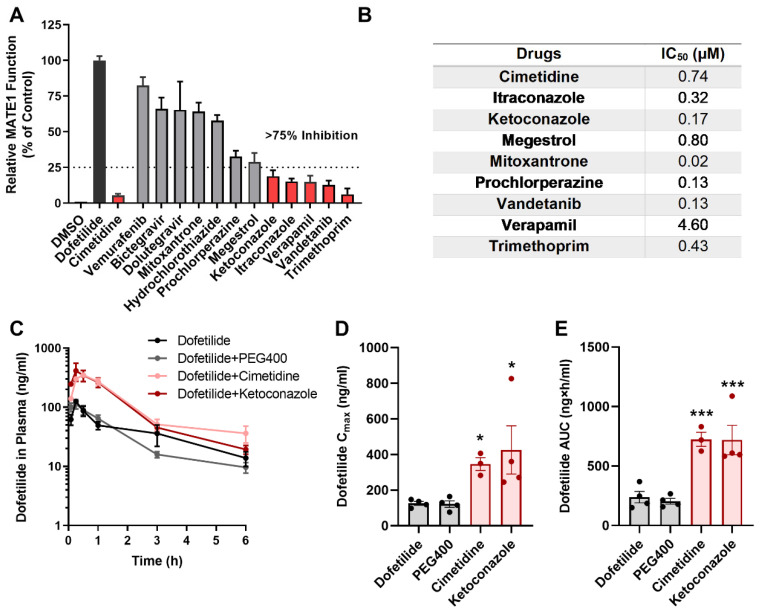
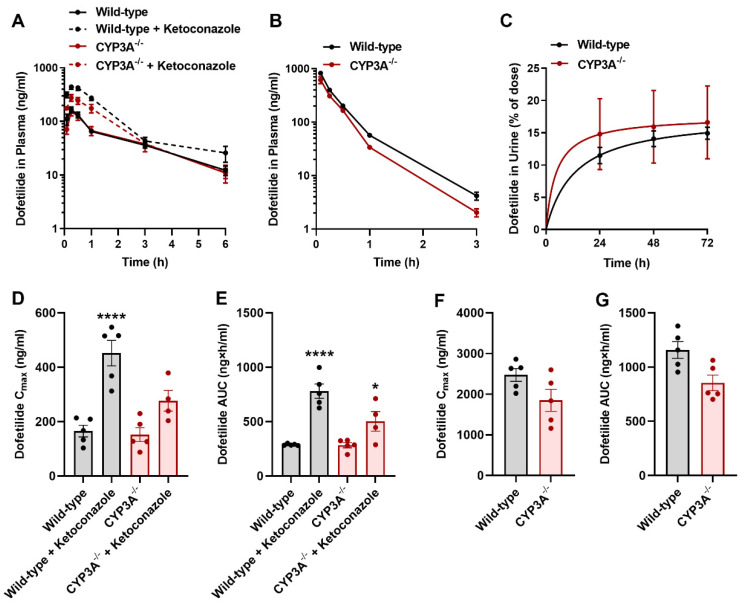
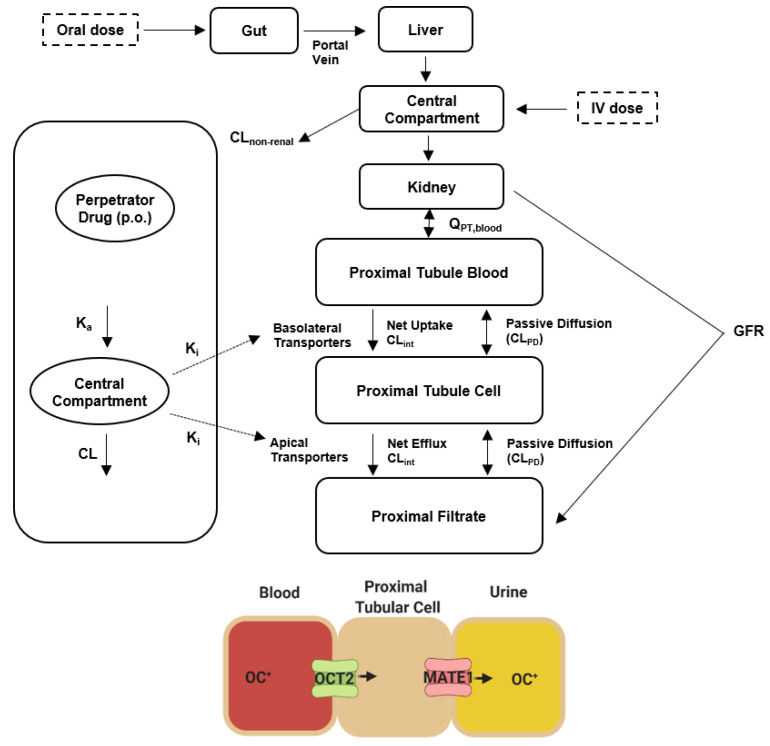
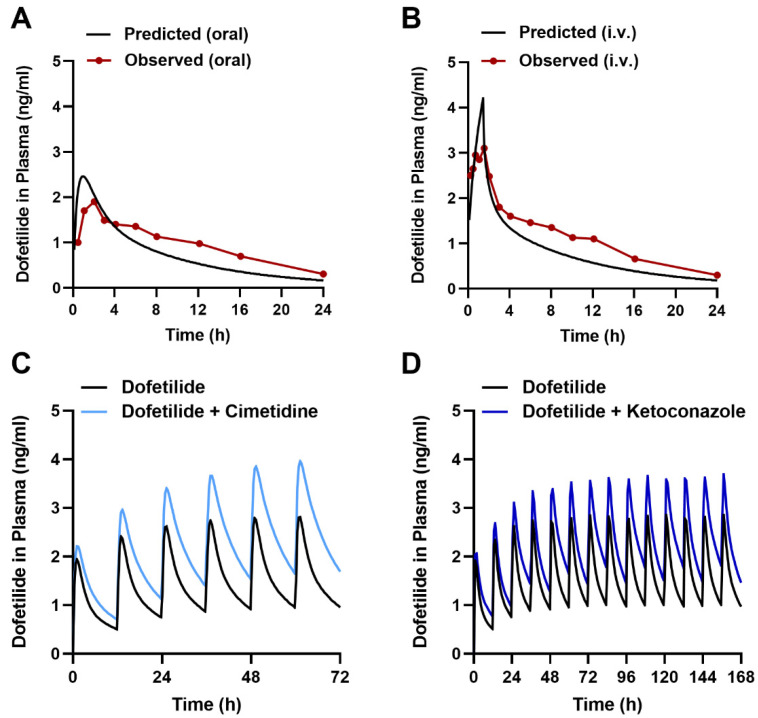
Dofetilide is a rapid delayed rectifier potassium current inhibitor widely used to prevent the recurrence of atrial fibrillation and flutter. The clinical use of this drug is associated with increases in QTc interval, which predispose patients to ventricular cardiac arrhythmias. The mechanisms involved in the disposition of dofetilide, including its movement in and out of cardiomyocytes, remain unknown. Using a xenobiotic transporter screen, we identified MATE1 (SLC47A1) as a transporter of dofetilide and found that genetic knockout or pharmacological inhibition of MATE1 in mice was associated with enhanced retention of dofetilide in cardiomyocytes and increased QTc prolongation. The urinary excretion of dofetilide was also dependent on the MATE1 genotype, and we found that this transport mechanism provides a mechanistic basis for previously recorded drug-drug interactions of dofetilide with various contraindicated drugs, including bictegravir, cimetidine, ketoconazole, and verapamil. The translational significance of these observations was examined with a physiologically-based pharmacokinetic model that adequately predicted the drug-drug interaction liabilities in humans. These findings support the thesis that MATE1 serves a conserved cardioprotective role by restricting excessive cellular accumulation and warrant caution against the concurrent administration of potent MATE1 inhibitors and cardiotoxic substrates with a narrow therapeutic window.
Keywords: PBPK modeling; arrhythmia; dofetilide; organic cation transporters.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Chugh S.S., Havmoeller R., Narayanan K., Singh D., Rienstra M., Benjamin E.J., Gillum R.F., Kim Y.H., McAnulty J.H., Zheng Z.J., et al. Worldwide Epidemiology of Atrial Fibrillation: A Global Burden of Disease 2010 Study. Circulation. 2014;129:837–847. doi: 10.1161/CIRCULATIONAHA.113.005119. - DOI - PMC - PubMed
-
- Krijthe B.P., Kunst A., Benjamin E.J., Lip G.Y., Franco O.H., Hofman A., Witteman J.C., Stricker B.H., Heeringa J. Projections on the Number of Individuals with Atrial Fibrillation in the European Union, from 2000 to 2060. Eur. Heart J. 2013;34:2746–2751. doi: 10.1093/eurheartj/eht280. - DOI - PMC - PubMed
-
- Alonso A., Krijthe B.P., Aspelund T., Stepas K.A., Pencina M.J., Moser C.B., Sinner M.F., Sotoodehnia N., Fontes J.D., Janssens A.C., et al. Simple Risk Model Predicts Incidence of Atrial Fibrillation in a Racially and Geographically Diverse Population: The CHARGE-AF Consortium. J. Am. Heart Assoc. 2013;2:e000102. doi: 10.1161/JAHA.112.000102. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
