

Inborn Errors of Nucleoside Transporter (NT)-Encoding Genes (SLC28 and SLC29)
- PMID: 35955904
- PMCID: PMC9369021
- DOI: 10.3390/ijms23158770
Inborn Errors of Nucleoside Transporter (NT)-Encoding Genes (SLC28 and SLC29)
Abstract
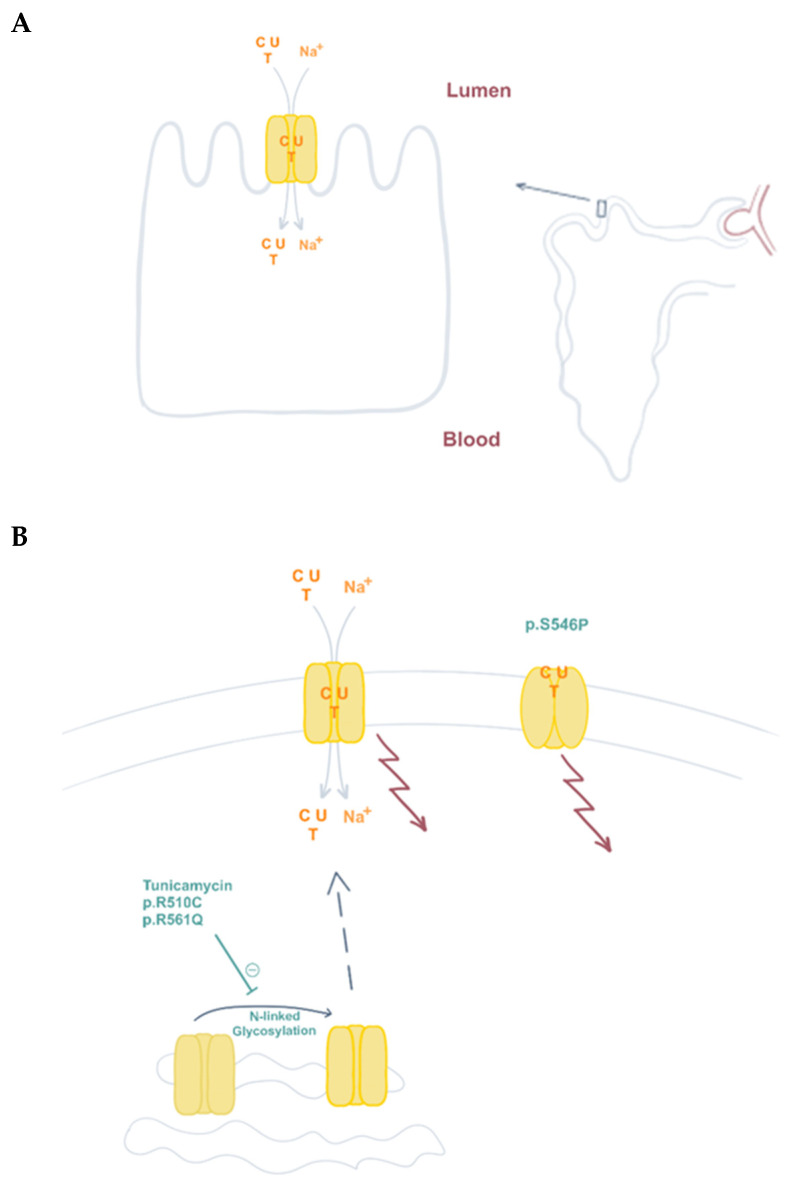
The proper regulation of nucleotide pools is essential for all types of cellular functions and depends on de novo nucleotide biosynthesis, salvage, and degradation pathways. Despite the apparent essentiality of these processes, a significant number of rare diseases associated with mutations in genes encoding various enzymes of these pathways have been already identified, and others are likely yet to come. However, knowledge on genetic alterations impacting on nucleoside and nucleobase transporters is still limited. At this moment three gene-encoding nucleoside and nucleobase transporter proteins have been reported to be mutated in humans, SLC29A1, SLC29A3, and SLC28A1, impacting on the expression and function of ENT1, ENT3, and CNT1, respectively. ENT1 alterations determine Augustine-null blood type and cause ectopic calcification during aging. ENT3 deficiency translates into various clinical manifestations and syndromes, altogether listed in the OMIM catalog as histiocytosis-lymphoadenopathy plus syndrome (OMIM#602782). CNT1 deficiency causes uridine-cytidineuria (URCTU) (OMIM#618477), a unique type of pyrimidineuria with an as yet not well-known clinical impact. Increasing knowledge on the physiological, molecular and structural features of these transporter proteins is helping us to better understand the biological basis behind the biochemical and clinical manifestations caused by these deficiencies. Moreover, they also support the view that some metabolic compensation might occur in these disturbances, because they do not seem to significantly impact nucleotide homeostasis, but rather other biological events associated with particular subtypes of transporter proteins.
Keywords: genetics; nucleosides; rare diseases; transporters.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Functional disruption of pyrimidine nucleoside transporter CNT1 results in a novel inborn error of metabolism with high excretion of uridine and cytidine.J Inherit Metab Dis. 2019 May;42(3):494-500. doi: 10.1002/jimd.12081. Epub 2019 Apr 8. J Inherit Metab Dis. 2019. PMID: 30847922
-
Equilibrative nucleotide transporter ENT3 (SLC29A3): A unique transporter for inherited disorders and cancers.Exp Cell Res. 2024 Jan 15;434(2):113892. doi: 10.1016/j.yexcr.2023.113892. Epub 2023 Dec 16. Exp Cell Res. 2024. PMID: 38104646 Review.
-
The concentrative nucleoside transporter family, SLC28.Pflugers Arch. 2004 Feb;447(5):728-34. doi: 10.1007/s00424-003-1107-y. Epub 2003 Jul 11. Pflugers Arch. 2004. PMID: 12856181 Review.
-
Transporters that translocate nucleosides and structural similar drugs: structural requirements for substrate recognition.Med Res Rev. 2012 Mar;32(2):428-57. doi: 10.1002/med.20221. Epub 2011 Feb 1. Med Res Rev. 2012. PMID: 21287570 Review.
-
Renal nucleoside transporters: physiological and clinical implications.Biochem Cell Biol. 2006 Dec;84(6):844-58. doi: 10.1139/o06-198. Biochem Cell Biol. 2006. PMID: 17215872 Review.
Cited by
-
Adenine-induced animal model of chronic kidney disease: current applications and future perspectives.Ren Fail. 2024 Dec;46(1):2336128. doi: 10.1080/0886022X.2024.2336128. Epub 2024 Apr 4. Ren Fail. 2024. PMID: 38575340 Free PMC article. Review.
-
Overcoming Biological Barriers: Importance of Membrane Transporters in Homeostasis, Disease and Disease Treatment.Int J Mol Sci. 2023 Apr 13;24(8):7212. doi: 10.3390/ijms24087212. Int J Mol Sci. 2023. PMID: 37108379 Free PMC article.
-
How Cryo-EM Has Expanded Our Understanding of Membrane Transporters.Drug Metab Dispos. 2023 Aug;51(8):904-922. doi: 10.1124/dmd.122.001004. Epub 2023 Jul 12. Drug Metab Dispos. 2023. PMID: 37438132 Free PMC article. Review.
-
Lysosomal dysfunction and overload of nucleosides in thymidine phosphorylase deficiency of MNGIE.J Transl Med. 2024 May 13;22(1):449. doi: 10.1186/s12967-024-05275-8. J Transl Med. 2024. PMID: 38741129 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- RTC2019-006865-1/Ministerio de Ciencia e Innovación, MCIN/ AEI /10.13039/501100011033/ FEDER "Una manera de hacer Europa"
- RTI2018-094655-B-I00/Ministerio de Ciencia e Innovación, MCIN/ AEI /10.13039/501100011033/ FEDER "Una manera de hacer Europa"
- RL001386/Fundació La Marató de TV3 - Rare diseases initiative
- G0063/Acción Estratégica CIBER EHD, Instituto de Salud Carlos III
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
