

Deletion of Meg8-DMR Enhances Migration and Invasion of MLTC-1 Depending on the CTCF Binding Sites
- PMID: 35955961
- PMCID: PMC9369160
- DOI: 10.3390/ijms23158828
Deletion of Meg8-DMR Enhances Migration and Invasion of MLTC-1 Depending on the CTCF Binding Sites
Abstract
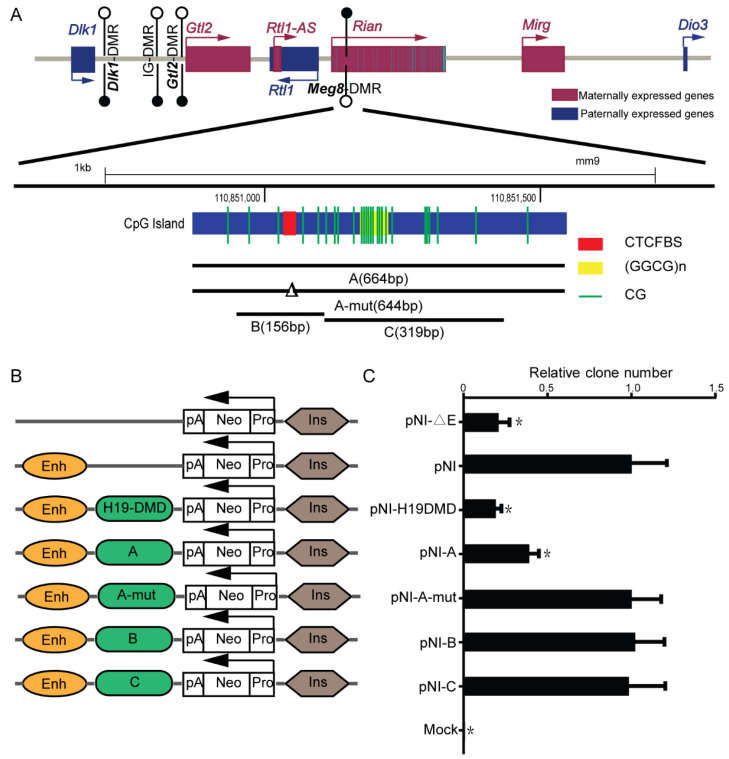
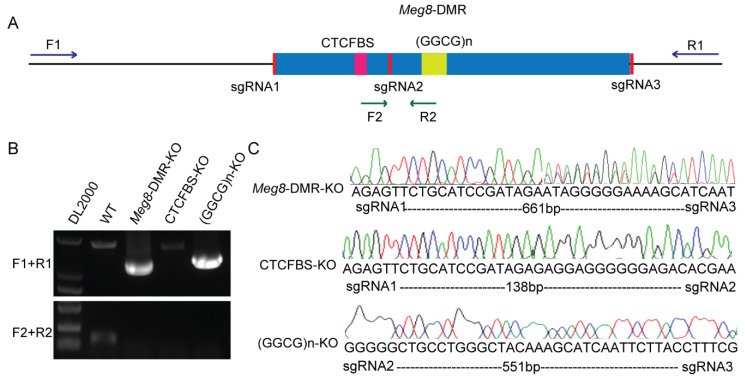
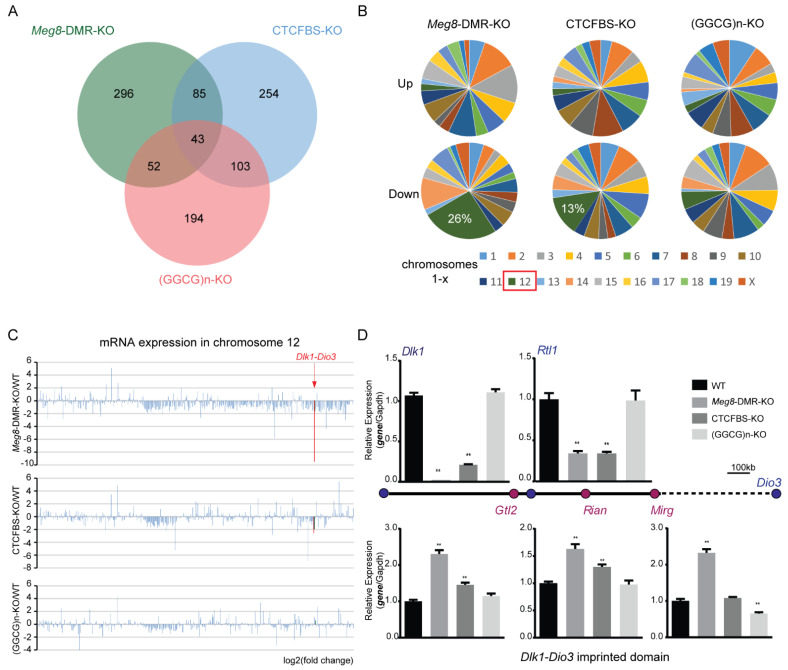
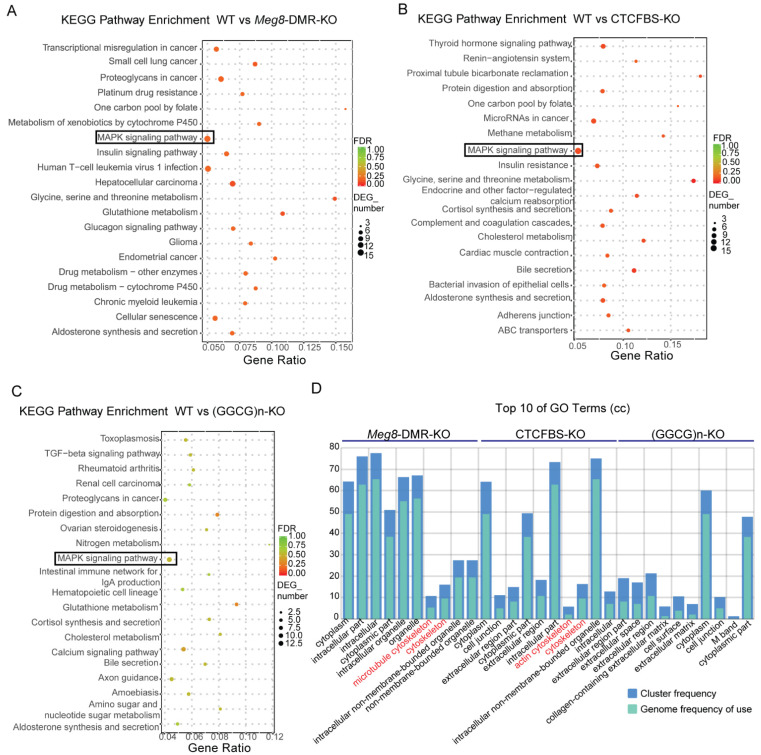
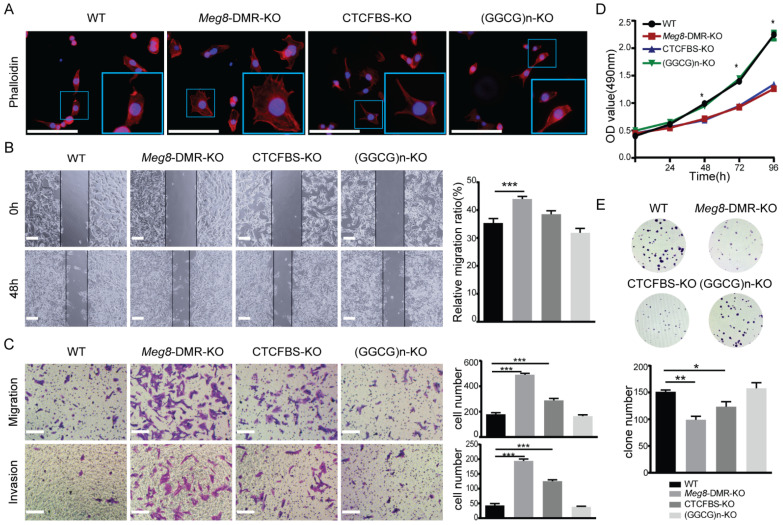
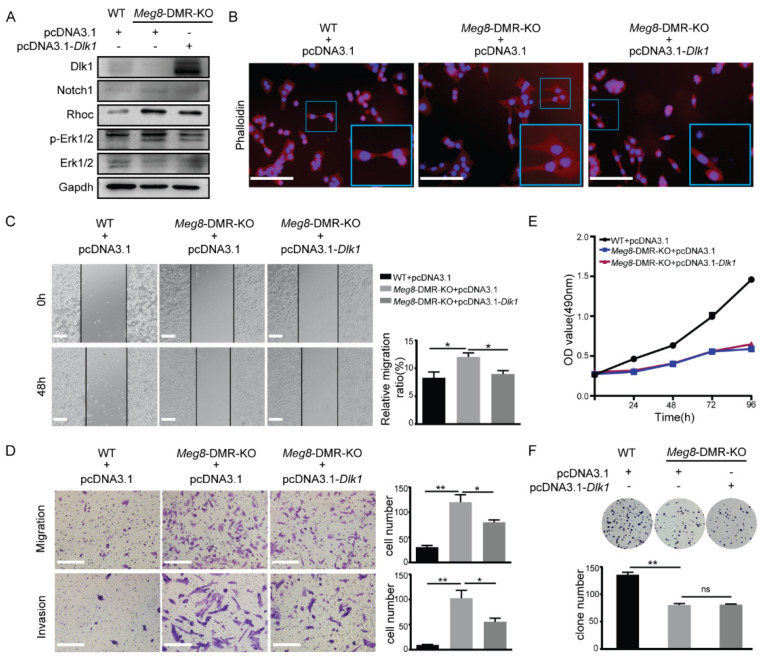
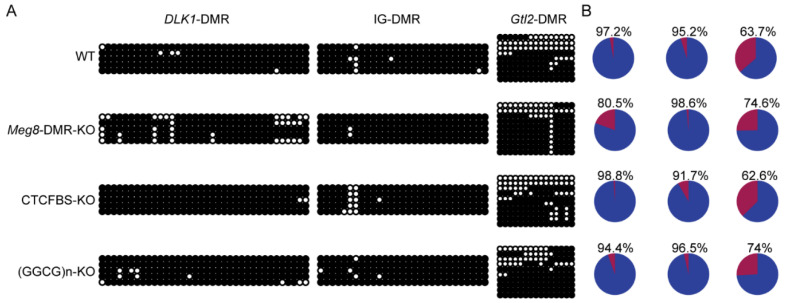
The Dlk1-Dio3 imprinted domain on mouse chromosome 12 contains three well-characterized paternally methylated differentially methylated regions (DMRs): IG-DMR, Gtl2-DMR, and Dlk1-DMR. These DMRs control the expression of many genes involved in embryonic development, inherited diseases, and human cancer in this domain. The first maternal methylation DMR discovered in this domain was the Meg8-DMR, the targets and biological function of which are still unknown. Here, using an enhancer-blocking assay, we first dissected the functional parts of the Meg8-DMR and showed that its insulator activity is dependent on the CCCTC-binding factor (CTCF) in MLTC-1. Results from RNA-seq showed that the deletion of the Meg8-DMR and its compartment CTCF binding sites, but not GGCG repeats, lead to the downregulation of numerous genes on chromosome 12, in particular the drastically reduced expression of Dlk1 and Rtl1 in the Dlk1-Dio3 domain, while differentially expressed genes are enriched in the MAPK pathway. In vitro assays revealed that the deletion of the Meg8-DMR and CTCF binding sites enhances cell migration and invasion by decreasing Dlk1 and activating the Notch1-Rhoc-MAPK/ERK pathway. These findings enhance research into gene regulation in the Dlk1-Dio3 domain by indicating that the Meg8-DMR functions as a long-range regulatory element which is dependent on CTCF binding sites and affects multiple genes in this domain.
Keywords: CTCF; Dlk1; Dlk1-Dio3 domain; MLTC-1; Meg8-DMR; invasion; migration.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Meg8-DMR as the Secondary Regulatory Region Regulates the Expression of MicroRNAs While It Does Not Affect Embryonic Development in Mice.Genes (Basel). 2023 Jun 14;14(6):1264. doi: 10.3390/genes14061264. Genes (Basel). 2023. PMID: 37372444 Free PMC article.
-
Novel 14q32.2 paternal deletion encompassing the whole DLK1 gene associated with Temple syndrome.Clin Epigenetics. 2024 May 7;16(1):62. doi: 10.1186/s13148-024-01652-8. Clin Epigenetics. 2024. PMID: 38715103 Free PMC article.
-
DNA methylation dynamics of a maternally methylated DMR in the mouse Dlk1-Dio3 domain.FEBS Lett. 2014 Dec 20;588(24):4665-71. doi: 10.1016/j.febslet.2014.10.038. Epub 2014 Nov 11. FEBS Lett. 2014. PMID: 25447521
-
Dysregulation of ncRNAs located at the DLK1‑DIO3 imprinted domain: involvement in urological cancers.Cancer Manag Res. 2019 Jan 15;11:777-787. doi: 10.2147/CMAR.S190764. eCollection 2019. Cancer Manag Res. 2019. PMID: 30697070 Free PMC article. Review.
-
[Review on the genomic imprinting at the mammalian DLK1-DIO3 cluster.].Yi Chuan. 2010 Aug;32(8):769-78. doi: 10.3724/sp.j.1005.2010.00769. Yi Chuan. 2010. PMID: 20709673 Review. Chinese.
Cited by
-
Meg8-DMR as the Secondary Regulatory Region Regulates the Expression of MicroRNAs While It Does Not Affect Embryonic Development in Mice.Genes (Basel). 2023 Jun 14;14(6):1264. doi: 10.3390/genes14061264. Genes (Basel). 2023. PMID: 37372444 Free PMC article.
-
Maternal RNA transcription in Dlk1-Dio3 domain is critical for proper development of the mouse placental vasculature.Commun Biol. 2024 Mar 23;7(1):363. doi: 10.1038/s42003-024-06038-3. Commun Biol. 2024. PMID: 38521877 Free PMC article.
-
Novel 14q32.2 paternal deletion encompassing the whole DLK1 gene associated with Temple syndrome.Clin Epigenetics. 2024 May 7;16(1):62. doi: 10.1186/s13148-024-01652-8. Clin Epigenetics. 2024. PMID: 38715103 Free PMC article.
-
Epigenetic control and genomic imprinting dynamics of the Dlk1-Dio3 domain.Front Cell Dev Biol. 2023 Dec 12;11:1328806. doi: 10.3389/fcell.2023.1328806. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 38155837 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
