

Occurrence of autoantibodies against skin proteins in patients with hereditary epidermolysis bullosa predisposes to development of autoimmune blistering disease
- PMID: 35958577
- PMCID: PMC9358991
- DOI: 10.3389/fimmu.2022.945176
Occurrence of autoantibodies against skin proteins in patients with hereditary epidermolysis bullosa predisposes to development of autoimmune blistering disease
Abstract
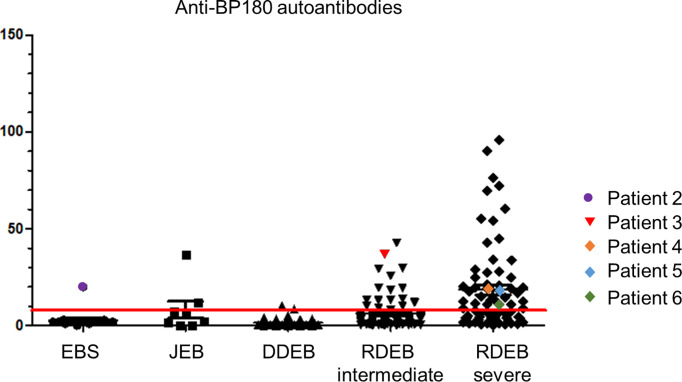
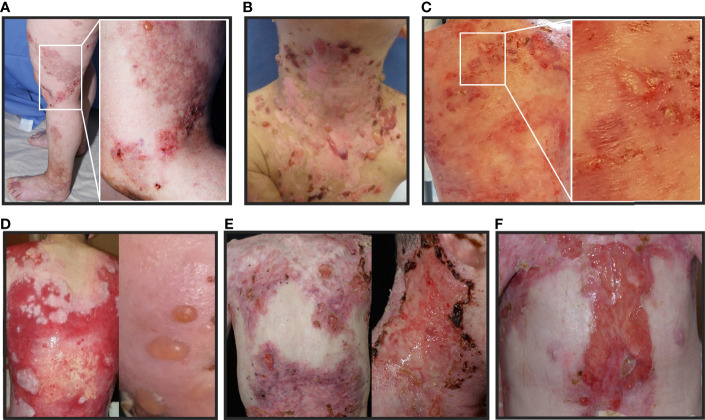
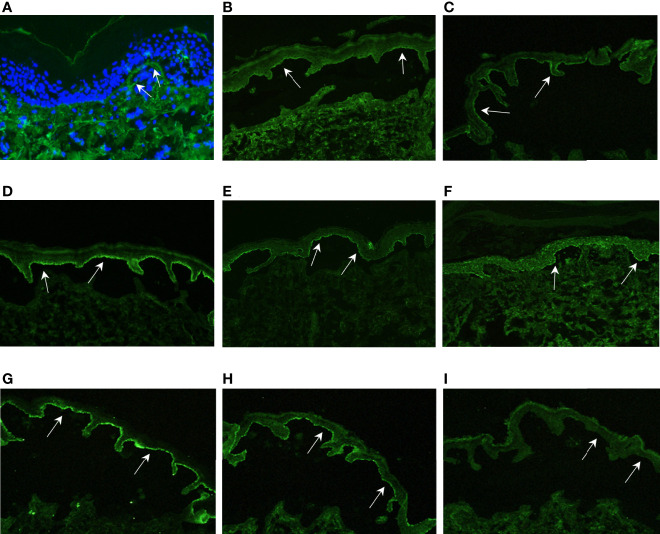
Skin blistering disorders are associated with inherited defects in proteins involved in the dermal-epidermal adhesion or autoantibodies targeting those proteins. Although blistering in hereditary epidermolysis bullosa (EB) is pathogenetically linked to genetic deficiency of distinct proteins of the epidermis or the dermal-epidermal junction, circulating autoantibodies against these proteins have also been identified in EB patients. So far, autoantibodies have been considered bystanders in EB and active pathogenicity of them in EB has not been disclosed. In sera of a cohort of 258 EB patients, we found by ELISA in 22% of the patients autoantibodies against the bullous pemphigoid antigen BP180. The titers correlated negatively with collagen VII skin expression and positively with disease severity. Among those patients, we identified six (2.33%) with clinical features of an autoimmune bullous disorder (AIBD) and positive indirect immunofluorescence (IIF) staining. In literature, we found four more cases of EB patients developing disease-aggravating AIBD. Co-existence of these two rare skin disorders suggests that EB patients have a predisposition for the development of AIBD. Our work highlights that EB patients with increased itch or blister formation should be evaluated for additional AIBD and repeated screening for changes in autoantibody titers and skin-binding specificities is advised.
Keywords: BP180; PD-1 inhibitor; autoimmune bullous disease; collagen VII; epidermolysis bullosa; skin blistering.
Copyright © 2022 Lehr, Felber, Tantcheva-Poór, Keßler, Eming, Nyström, Rizzi and Kiritsi.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Patients suffering from dystrophic epidermolysis bullosa are prone to developing autoantibodies against skin proteins: A longitudinal confirmational study.Exp Dermatol. 2024 Feb;33(2):e15035. doi: 10.1111/exd.15035. Exp Dermatol. 2024. PMID: 38389191
-
Prevalence of specific anti-skin autoantibodies in a cohort of patients with inherited epidermolysis bullosa.Orphanet J Rare Dis. 2013 Sep 4;8:132. doi: 10.1186/1750-1172-8-132. Orphanet J Rare Dis. 2013. PMID: 24007552 Free PMC article.
-
Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid.J Pathol. 2004 Dec;204(5):519-27. doi: 10.1002/path.1674. J Pathol. 2004. PMID: 15538734
-
Autoimmune Subepidermal Bullous Diseases of the Skin and Mucosae: Clinical Features, Diagnosis, and Management.Clin Rev Allergy Immunol. 2018 Feb;54(1):26-51. doi: 10.1007/s12016-017-8633-4. Clin Rev Allergy Immunol. 2018. PMID: 28779299 Review.
-
A Review of Acquired Autoimmune Blistering Diseases in Inherited Epidermolysis Bullosa: Implications for the Future of Gene Therapy.Antibodies (Basel). 2021 May 17;10(2):19. doi: 10.3390/antib10020019. Antibodies (Basel). 2021. PMID: 34067512 Free PMC article. Review.
Cited by
-
Unveiling the value of C-reactive protein as a severity biomarker and the IL4/IL13 pathway as a therapeutic target in recessive dystrophic epidermolysis bullosa: A multiparametric cross-sectional study.Exp Dermatol. 2024 Aug;33(8):e15146. doi: 10.1111/exd.15146. Exp Dermatol. 2024. PMID: 39075828 Free PMC article.
-
Skin Deep and Beyond: Unravelling B Cell Extracellular Matrix Interactions in Cutaneous Immunity and Disease.Exp Dermatol. 2025 Mar;34(3):e70068. doi: 10.1111/exd.70068. Exp Dermatol. 2025. PMID: 40051023 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials