

SERPING1 Variants and C1-INH Biological Function: A Close Relationship With C1-INH-HAE
- PMID: 35958943
- PMCID: PMC9361472
- DOI: 10.3389/falgy.2022.835503
SERPING1 Variants and C1-INH Biological Function: A Close Relationship With C1-INH-HAE
Abstract
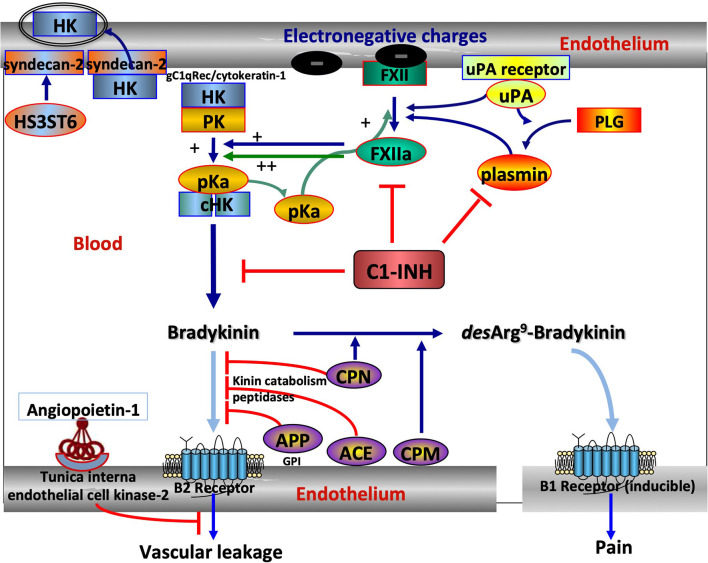
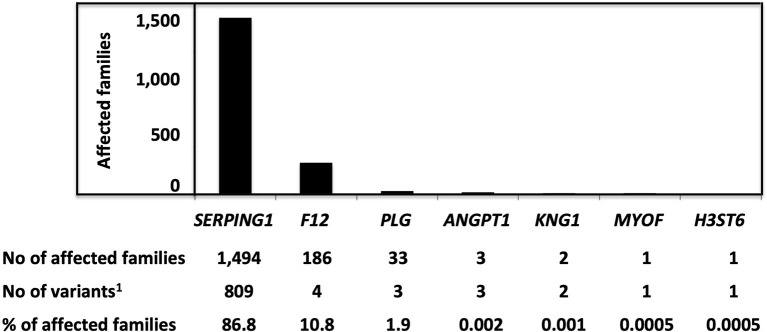
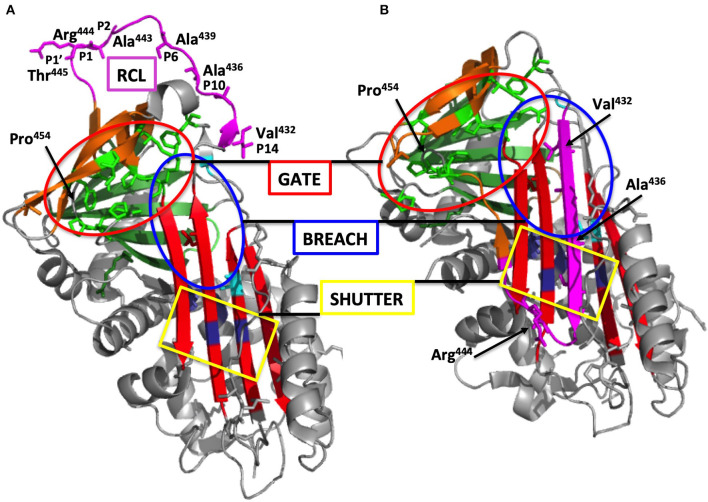
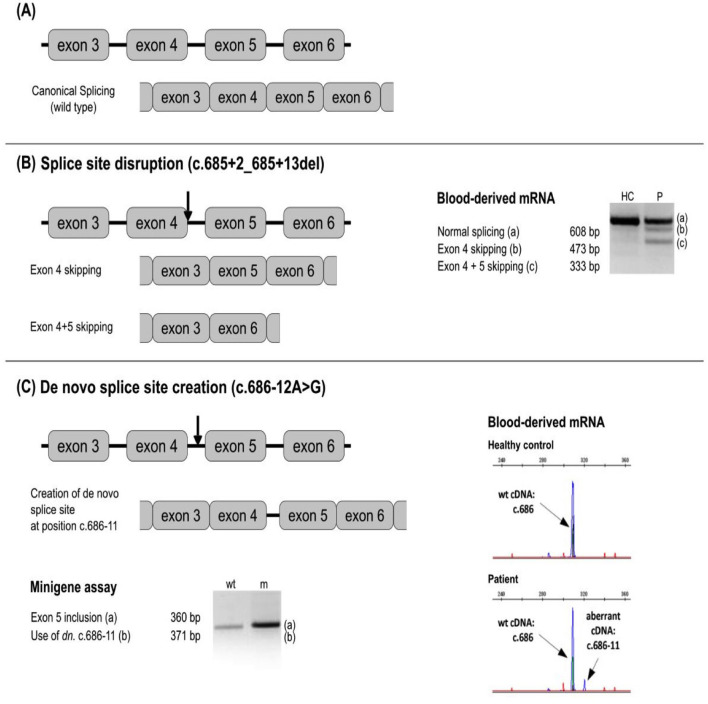
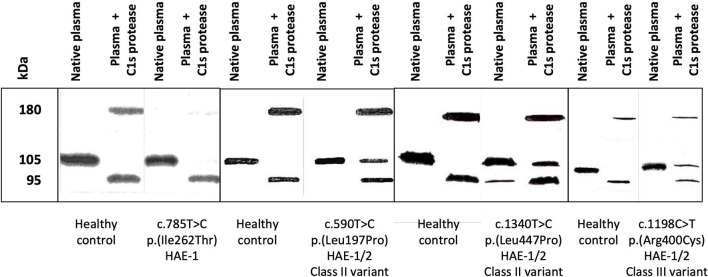
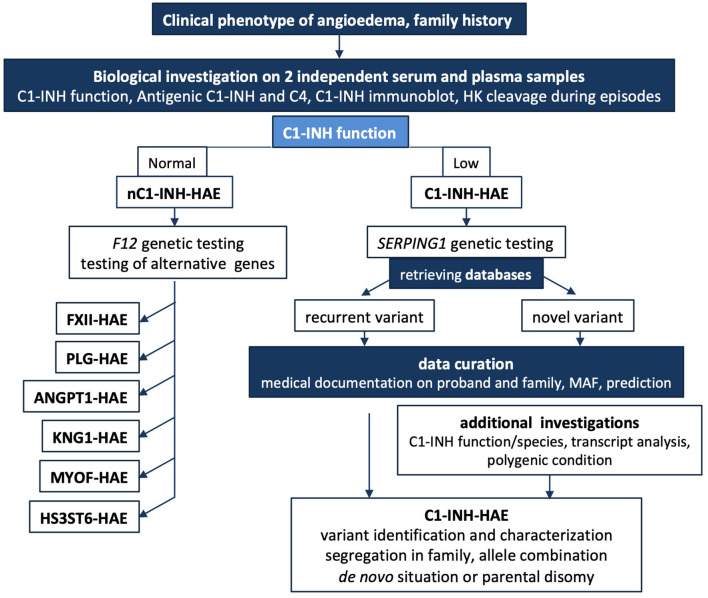
Hereditary angioedema with C1 Inhibitor deficiency (C1-INH-HAE) is caused by a constellation of variants of the SERPING1 gene (n = 809; 1,494 pedigrees), accounting for 86.8% of HAE families, showing a pronounced mutagenic liability of SERPING1 and pertaining to 5.6% de novo variants. C1-INH is the major control serpin of the kallikrein-kinin system (KKS). In addition, C1-INH controls complement C1 and plasminogen activation, both systems contributing to inflammation. Recognizing the failed control of C1s protease or KKS provides the diagnosis of C1-INH-HAE. SERPING1 variants usually behave in an autosomal-dominant character with an incomplete penetrance and a low prevalence. A great majority of variants (809/893; 90.5%) that were introduced into online database have been considered as pathogenic/likely pathogenic. Haploinsufficiency is a common feature in C1-INH-HAE where a dominant-negative variant product impacts the wild-type allele and renders it inactive. Small (36.2%) and large (8.3%) deletions/duplications are common, with exon 4 as the most affected one. Point substitutions with missense variants (32.2%) are of interest for the serpin structure-function relationship. Canonical splice sites can be affected by variants within introns and exons also (14.3%). For noncanonical sequences, exon skipping has been confirmed by splicing analyses of patients' blood-derived RNAs (n = 25). Exonic variants (n = 6) can affect exon splicing. Rare deep-intron variants (n = 6), putatively acting as pseudo-exon activating mutations, have been characterized as pathogenic. Some variants have been characterized as benign/likely benign/of uncertain significance (n = 74). This category includes some homozygous (n = 10) or compound heterozygous variants (n = 11). They are presenting with minor allele frequency (MAF) below 0.00002 (i.e., lower than C1-INH-HAE frequency), and may be quantitatively unable to cause haploinsufficiency. Rare benign variants could contribute as disease modifiers. Gonadal mosaicism in C1-INH-HAE is rare and must be distinguished from a de novo variant. Situations with paternal or maternal disomy have been recorded (n = 3). Genotypes must be interpreted with biological investigation fitting with C1-INH expression and typing. Any SERPING1 variant reminiscent of the dysfunctional phenotype of serpin with multimerization or latency should be identified as serpinopathy.
Keywords: C1 Inhibitor; C1-INH-HAE; SERPING1 gene; angioedema; genetic variation; hereditary–diagnosis; serpin function; serpinopathy.
Copyright © 2022 Drouet, López-Lera, Ghannam, López-Trascasa, Cichon, Ponard, Parsopoulou, Grombirikova, Freiberger, Rijavec, Veronez, Pesquero and Germenis.
Conflict of interest statement
AGh was employed by KininX SAS. FP and AGe were employed by CeMIA SA. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous