

Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure
- PMID: 35959412
- PMCID: PMC9364913
- DOI: 10.1038/s44161-022-00028-6
Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure
Abstract
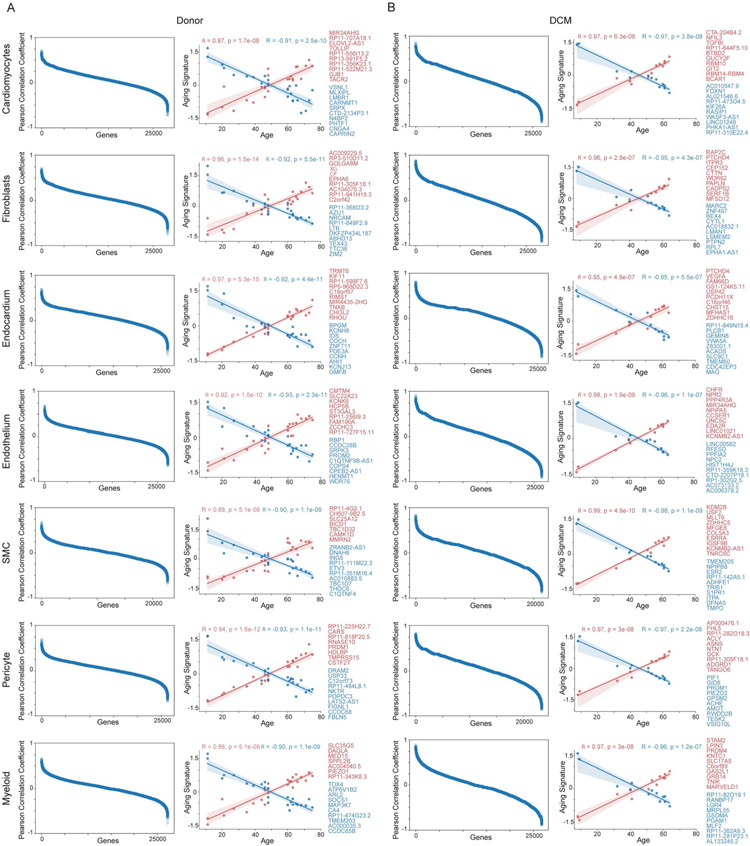
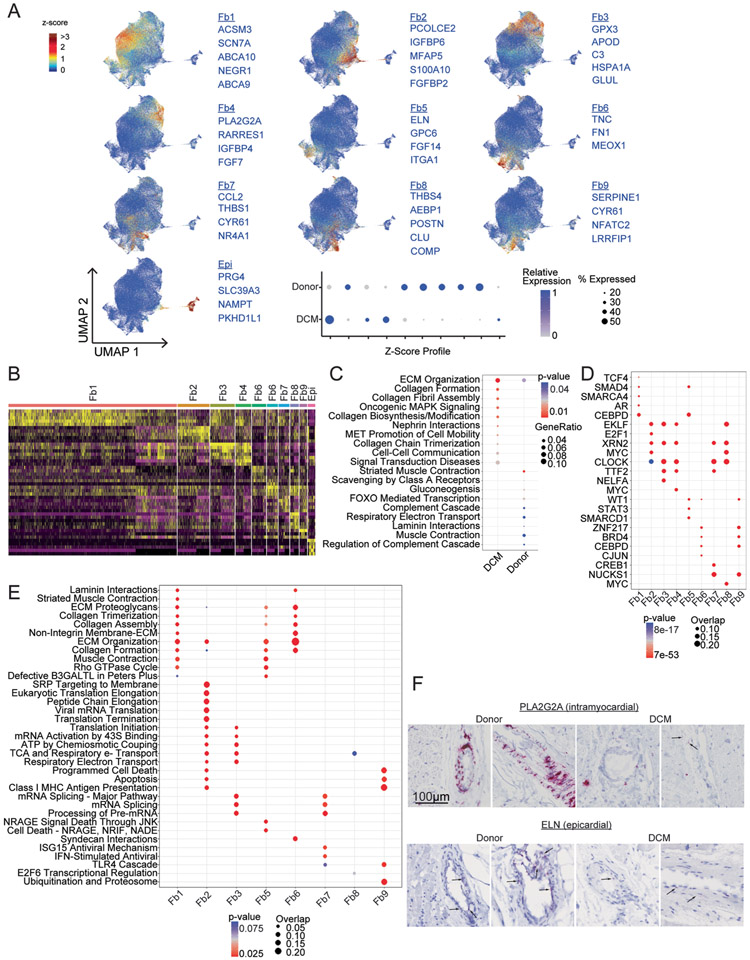
Heart failure represents a major cause of morbidity and mortality worldwide. Single-cell transcriptomics have revolutionized our understanding of cell composition and associated gene expression. Through integrated analysis of single-cell and single-nucleus RNA-sequencing data generated from 27 healthy donors and 18 individuals with dilated cardiomyopathy, here we define the cell composition of the healthy and failing human heart. We identify cell-specific transcriptional signatures associated with age and heart failure and reveal the emergence of disease-associated cell states. Notably, cardiomyocytes converge toward common disease-associated cell states, whereas fibroblasts and myeloid cells undergo dramatic diversification. Endothelial cells and pericytes display global transcriptional shifts without changes in cell complexity. Collectively, our findings provide a comprehensive analysis of the cellular and transcriptomic landscape of human heart failure, identify cell type-specific transcriptional programs and disease-associated cell states and establish a valuable resource for the investigation of human heart failure.
Figures
















References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases